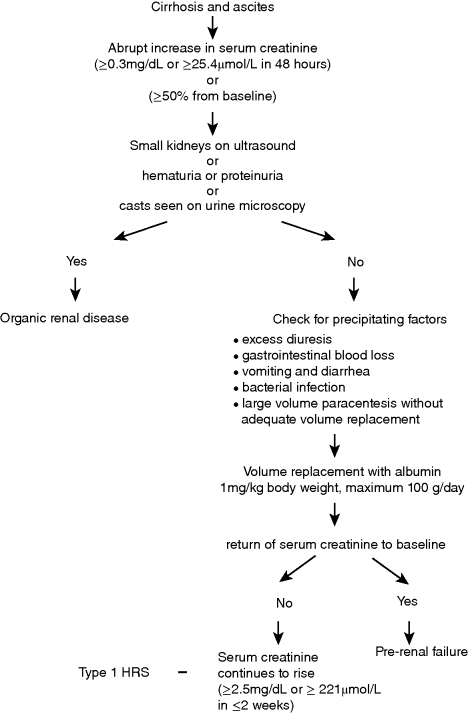
Chapter 16 Florence Wong Division of Gastroenterology, Department of Medicine, Toronto General Hospital, University of Toronto Ontario Canada Renal dysfunction is a common complication of liver cirrhosis, estimated to occur in approximately 20% of patients with advanced cirrhosis and ascites admitted to hospital [1]. Although the prevalence of chronic kidney disease (CKD) is rising, related to an increasing number of patients worldwide affected by viral hepatitis, obesity, diabetes, and hypertension, the majority of cases of renal dysfunction are related to acute kidney injury (AKI), whether occurring de novo or superimposed on a background of CKD [1,2]. While structural renal disease can occur in patients with cirrhosis, leading to acute renal dysfunction, most of the AKI cases in cirrhosis are functional in nature, that is, there is no underlying structural abnormality in the kidneys; rather, the renal dysfunction is the result of hemodynamic changes that are prevalent in advanced cirrhosis. There is splanchnic and systemic arterial vasodilatation, which leads to an apparent decrease in the intravascular volume, the so-called reduction in the effective arterial blood volume. The physiologic response is an activation of various vasoconstrictor systems in an attempt to reduce the extent of vasodilatation, and to improve the intravascular volume by increasing renal sodium and water retention [3]. The renal circulation is very sensitive to the vasoconstrictive effects of these vasoconstrictor systems and therefore is susceptible to the development of renal vasoconstriction with renal impairment. Most episodes of functional renal disorder are responsive to intravascular volume replacement, with return of the renal function to the baseline level [1]. Hepatorenal syndrome (HRS) is the most severe form of functional renal failure and it is not responsive to intravascular volume challenge. The exact prevalence of AKI is unknown, as many cases of mild AKI are outpatients and therefore may escape medical attention. Amongst hospitalized patients, AKI including cases of structural renal disease and functional renal failure, has been reported to have a prevalence of 19% [1]. In a series of 263 cirrhotic patients with ascites admitted to two large tertiary centers in Barcelona for management of ascites, 27.4% of patients developed prerenal renal failure (volume-responsive functional renal failure, FRF), 14.1% developed infection-related FRF, and 7.6% developed HRS during a mean follow-up period of 41 months [4]. Interestingly, 7.6% of patients also developed renal failure due to structural renal diseases during follow-up [4]. Because the development of AKI in cirrhosis is associated with increased morbidity and mortality, even before patients reach the stage of established HRS [5,6], significant efforts have been made in recent years to better our understanding of the pathophysiology, and to update the various definitions and diagnostic criteria. HRS is the most clearly defined and best studied amongst all the AKI entities in cirrhosis. It used to be an invariably fatal condition with a median survival of several days without treatment [7]. With the introduction of effective treatments, HRS is no longer a terminal condition. Therefore, it is paramount that early and accurate diagnosis is made, so as to implement treatment to improve the outcome for these patients. In 2007, the International Ascites Club, an organization consisting of experts in hepatology, renal physiology, hemodynamics, electrolyte disorders, cardiopulmonary function, and transplant medicine, revised the definition of HRS. The group wanted to emphasize the functional nature of the condition, which is consequent upon the systemic vasodilatory hemodynamic changes that occur in advanced cirrhosis [8–10]. The group also recognized that changes in cardiac function and renal physiology contributed to the development of HRS (Table 16.1). For example, patients who developed renal failure with an episode of bacterial peritonitis had significantly lower cardiac output at the time of infection resolution when compared with baseline, and also when compared with those who did not develop HRS at the time of infection resolution [11]. Changes in renal autoregulation with progression of liver cirrhosis also has a role in the development of HRS. Autoregulation is a physiologic response that maintains renal blood flow despite alterations in renal perfusion pressure. However, increasing levels of sympathetic nervous activity with advancement of cirrhosis can lead to progressive reduction in renal blood flow even with adequate renal perfusion pressure, thus predisposing the patient to the development of renal failure [12]. Table 16.1 Pathophysiologic processes that have been implicated in the development of hepatorenal syndrome (HRS). Thus, the current definition of HRS states that: HRS is a potentially reversible syndrome occurring in patients with cirrhosis, ascites and liver failure. It is characterized by impaired renal function, marked alterations in the cardiovascular function and over-activity of the endogenous vasoactive systems. Marked vasoconstriction in the kidney causes low glomerular filtration, whereas in the systemic circulation, there is decreased vascular resistance due to splanchnic and peripheral arterial vasodilatation. A similar syndrome can also occur in acute liver failure and acute alcoholic hepatitis. [13] A recent report has confirmed that patients with lower cardiac output had worse renal function as indicated by a lower glomerular filtration rate, lower renal blood flow, and higher serum creatinine, indicating a link between cardiac function and renal function [14]. Furthermore, those who went on to develop HRS had a lower cardiac index, as did those who died from their HRS [14]. Indeed, a cardiorenal interaction has been proposed in patients with advanced cirrhosis where cardiac dysfunction in cirrhosis is a major determinant of kidney function [15]. Therefore, there may be further refinement of the definition in the future as new pathophysiologic processes are recognized. Table 16.2 sets out the most recent criteria for the diagnosis of HRS, which continues to mandate a diagnosis of exclusion [13]. These diagnostic criteria have been simplified to improve the diagnostic accuracy of HRS, thereby allowing application of effective treatment options according to a standard set of rules. Of note, HRS can be diagnosed in the setting of a bacterial infection, provided septic shock is not present. In addition, urinary electrolyte or creatinine clearance is not required for the diagnosis of HRS, thereby obviating the need for a 24-hour urine collection. Despite its limitations as an accurate index of renal function [16,17], especially in advanced cirrhosis, serum creatinine is still the parameter used to diagnose HRS, because it is widely available and easy to measure. The diagnosis of HRS requires a serum creatinine level of >1.5 mg/dL (133 μmol/L). Clinically, HRS can be divided into two types: the acute form or type 1 HRS which is characterized by a rapidly progressive renal failure defined by doubling of the initial serum creatinine to a level greater than 2.5 mg/dL (221 μmol/L) in less than 2 weeks; whereas the chronic form or type 2 HRS is characterized by moderate renal failure (serum creatinine of 1.5–2.5 mg/dL or 133–221 μmol/L), which follows a steady or slowly progressive course, often associated with refractory ascites. Despite the emergence of several promising markers of renal dysfunction such as cystatin C, neutrophil gelatinase-associated lipocalin, interleukin-18, and liver type fatty acid binding protein [18], there are as yet no specific diagnostic markers for HRS. Many of these biomarkers are more specific for renal tubular damage and are therefore more appropriate for separating acute tubular necrosis from functional renal disorders rather than diagnosing hepatorenal syndrome per se. Table 16.2 Diagnostic criteria for hepatorenal syndrome. Source: Adapted from Salerno et al., 2007 [13]. Reproduced with permision of BMJ Group. Despite setting down the diagnostic criteria, the accurate diagnosis of HRS may not always be easy in daily clinical practice [19]. This is especially true for patients with rapidly deteriorating renal dysfunction, many of whom may be oliguric or anuric, thus making exclusion of structural renal disease on urinary criteria difficult. The volume replacement may not have followed strictly the criteria required for the diagnosis of HRS or the diuretic withdrawal may not have been complete before starting definitive treatment for HRS, as many physicians are unwilling to take a wait-and-see approach until the diagnostic serum creatinine level of >2.5 mg/dL (221 μmol/L) is reached. In a recent report of 263 consecutive patients with cirrhosis and renal failure admitted into 21 Italian hospitals, the cases of presumed diagnosis of HRS responded to treatment with vasoconstrictors with similar outcomes when compared with patients who fulfilled all the diagnostic criteria for HRS [19]. Therefore, physicians are encouraged to follow the diagnostic criteria for HRS, but should not delay or withhold treatment in cases of presumed HRS diagnosed on clinical grounds. Figure 16.1 sets out the diagnostic approach to a patient with cirrhosis, ascites, and renal dysfunction. Figure 16.1 Approach to a patient with cirrhosis and ascites who presents with an acute rise in serum creatinine fulfilling the diagnostic criteria for acute kidney injury. The most frequent differential diagnosis for HRS is acute tubular necrosis (ATN) and differentiating the two conditions is not always easy. Patients with ATN may not always provide a history of hypotension, or nephrotoxic drug use. Likewise, patients with a prolonged course of HRS may also develop ATN, related to intense renal arteriolar vasoconstriction, causing tubular damage [20]. Granular casts may be seen in both HRS and ATN. Urinary parameters such as urinary sodium excretion and urinary: plasma osmolality ratio are of no value in differentiating the two conditions [21]. The fact that patients who had abnormal serum creatinine but no proteinuria nor hematuria all showed structural renal changes on transvenous renal biopsy further adds to the confusion [22]. The recent report of the differential capability of various renal biomarkers holds promise in separating ATN from HRS [23], but this needs to be confirmed. Until then, physicians who lean towards the diagnosis of HRS should institute treatment for HRS, as renal dysfunction will invariably progress while waiting for clear differentiation, thus diminishing the likelihood of response to treatment [24]. Type 1 HRS is usually part of a multi-organ failure picture of the acute-on-chronic liver failure syndrome in a very ill patient with advanced cirrhosis [25]. Although type 1 HRS can develop spontaneously, it usually is precipitated by an acute event that perturbs the already precarious hemodynamic state of the patient. Therefore, treatment for type 1 HRS involves managing the precipitating event, the associated systemic inflammatory response syndrome, and the renal failure as well as the global organ malfunction of the patient. The most common precipitating event for type 1 HRS is bacterial infection [26]. The myriads of inflammatory cytokines released by the bacteria can induce various changes in the microcirculation including microthrombi in capillaries, endothelial dysfunction, and capillary leaks in many organs including the kidneys, leading to organ dysfunction [27]. Although spontaneous bacterial peritonitis is the most frequent precipitating event for type 1 HRS [26,28], all bacterial infections can cause renal failure [29]. In addition, any event that disturbs the volume status or exaggerates the reduction of intravascular volume such as gastrointestinal blood loss, overdiuresis, or large-volume paracentesis without adequate intravascular volume replacement can also precipitate type 1 HRS. Given that type 1 HRS is potentially reversible, it is imperative that the precipitating event is recognized early and treated accordingly. Volume replacement in the form of blood or blood products is given in cases of blood or blood volume loss. Diuretics and nephrotoxic drugs need to be withdrawn. In patients suspected of having an episode of bacterial infection, early and empirical use of antibiotics is recommended [30]. In those patients who have one of these precipitating events and who have not yet developed type 1 HRS, preventative measures can sometimes obviate its occurrence. For example, the incidence of renal impairment is most significant among patients with spontaneous bacterial peritonitis and a serum bilirubin level >4 mg/dL (68 μmol/L) and a serum creatinine level >1 mg/dL (88 μmol/L) [31], and prophylactic use of albumin in this scenario has been shown to reduce the incidence of renal failure [31]. Likewise, the use of prophylactic antibiotics in patients who have an episode of gastrointestinal bleeding has been shown to reduce the incidence of bacterial infections and therefore the likelihood of developing type 1 HRS [32]. Given that one of the major pathophysiologic factors for the development of type 1 HRS is systemic arterial vasodilatation, leading to paradoxical renal vasoconstriction, it stands to reason to treat type 1 HRS by counteracting the systemic arterial vasodilatation. The resultant improvement in the effective arterial blood volume should lead to a dampening of the activities of the various systemic vasoconstrictors, thereby decreasing the renal vasoconstriction, with overall improvement in renal blood flow and glomerular filtration. The most widely used agent outside North America for the treatment of type 1 HRS is terlipressin, which is a vasopressin analog. Terlipressin binds to the V1 receptors of the vascular smooth muscle cells to cause vasoconstriction in both the systemic and the splanchnic circulations [33]. It therefore reduces the extent of splanchnic and systemic vasodilatation. In addition, terlipressin has been shown to dilate intrahepatic vessels, thereby reducing intrahepatic resistance to portal inflow [34]. The overall result is a reduction in portal pressure, which also has a direct effect on improving renal function [35]. To date, there have been two randomized controlled trials comparing terlipressin and albumin with albumin alone [36], or terlipressin and albumin with placebo [37]. Both studies have shown efficacy in reversing type 1 HRS in approximately 34–43% of patients, but survival was not improved. Recurrence of HRS was not common after discontinuation of treatment, and any recurrence could be retreated with terlipressin with similar response as the initial course. Sub-analyses showed that patients with severe liver or renal dysfunction, as indicated by a serum bilirubin of >10 mg/dL (170 μmol/L) and a serum creatinine of >5.6 mg/dL (493 μmol/L) [24] were not likely to respond. In addition, those patients who did not have an increase in mean arterial pressure of >5 mmHg on day 3 were also likely to not respond to treatment [38]. The corollary from these observations is that patients who are very ill with severe liver and renal dysfunction should not be treated with terlipressin, and, for those who receive terlipressin, a lack of hemodynamic response on day 3 should also indicate that an alternative therapy should be given. More recently, there are reports that a continuous infusion is superior to bolus doses of terlipressin, which were used in the randomized controlled trials in the management of type 1 HRS [39,40]. Equal efficacy was achieved with a lower total daily dose and with fewer side effects, due to a more sustained portal pressure lowering effect of a continuous infusion than with bolus injections. There are now also case reports documenting the efficacy of repeated courses of continuous infusions of terlipressin with albumin 20–40 g/day as a bridge therapy to liver transplantation for repeated recurrences of HRS [41,42]. All patients who were treated in this manner remained alive at 46 days to 8 months and ultimately received a liver transplant as definitive therapy. Therefore, long-term use of terlipressin is a plausible option, preventing irreversible renal failure and the need for dialysis while waiting for liver transplantation. Midodrine is an α-agonist. It acts by binding to the α1
Hepatorenal Syndrome and Acute Kidney Injury
Hepatorenal Syndrome
Definition
Pathophysiologic process
Underlying etiology
Consequence
Splanchnic and systemic arterial vasodilatation [3]
Cardiac dysfunction
Altered renal autoregulation
Diagnostic criteria
Differential Diagnosis of Renal Dysfunction in Cirrhosis
Treatment of Type 1 Hepatorenal Syndrome
Treatment of Precipitating Event
Vasoconstrictor Therapy
Terlipressin
Midodrine
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel







Full access? Get Clinical Tree


