CHAPTER 79 Hepatitis C
Hepatitis C virus (HCV) infects 170 million people worldwide and 1.6% of the population of the United States.1–4 Unfortunately, HCV successfully evades the host immune response in 55% to 85% of acutely infected persons, thus leading to chronic infection. The natural history of hepatitis C varies greatly; reasons for this heterogeneity remain incompletely understood but are related to both viral, host, and environmental factors. Chronic HCV infection can lead to cirrhosis and hepatocellular carcinoma. The incidence of these complications has risen dramatically in the 2000s but is expected to decline over the next 20 years.3 In fact, whereas HCV-related mortality increased dramatically after 1995, it has reached a plateau since 2002.5 Complications of HCV-related cirrhosis are currently the leading indication for liver transplantation in the United States and Europe.
Chronic hepatitis C is the only chronic viral infection that can be cured by antiviral therapy. Currently, 40% to 50% of patients infected with HCV genotype 1 who tolerate full-dose treatment with pegylated interferon and ribavirin achieve a sustained virologic response (SVR) to treatment, defined as absence of HCV RNA in serum six months after discontinuation of treatment; an SVR is almost always associated with a durable eradication of the virus.6,7 From 70% to 80% of genotype 2- and 3-infected patients achieve an SVR. Substantial progress in understanding the mechanisms of virus entry into the hepatocyte, replication, and the host immune response has led to the development of new therapeutic agents that target the steps in the viral life cycle. New agents under investigation promise to improve the SVR further when combined with interferons and ribavirin.
VIROLOGY
STRUCTURE
The HCV virion has been visualized by electron microscopy and is an enveloped virus 50 nm in diameter.8 The two envelope proteins, E1 and E2, heterodimerize and assemble into tetramers, which create a smooth outer layer. This layer has a “fishbone” configuration with icosahedral symmetry. The envelope proteins are anchored to a host cell–derived lipid bilayer envelope membrane that surrounds the nucleocapsid. The nucleocapsid is believed to be composed of multiple copies of the core protein and forms an internal icosahedral viral coat that encapsulates the genomic ribonucleic acid (RNA). HCV circulates in various forms in the serum of an infected host, including (1) virions that are bound to very-low-density and low-density lipoproteins and appear to represent the infectious fraction; (2) virions bound to immunoglobulins; and (3) free virions. In addition, viral particles that exhibit physicochemical, morphologic, and antigenic properties of nonenveloped HCV nucleocapsids have been detected in plasma.9
GENOMIC ORGANIZATION
HCV is a single-stranded positive-sense RNA virus that belongs to the Flaviviridae family and has been classified as the sole member of the genus Hepacivirus.10 The genome of HCV contains approximately 9600 nucleotides with an open reading frame (ORF) that encodes one large viral polypeptide precursor of 3008 to 3033 amino acids. The HCV ORF is flanked upstream by a 5′ untranslated region (UTR) that functions as an internal ribosome entry site (IRES) to direct cap-independent translation (i.e., without the addition of an extra ribonucleotide to the 5′ end of the viral messenger RNA) and downstream by a 3′ UTR that is critical for initiation of new RNA strand synthesis.11–13 The 5′ and portions of the 3′ UTR are the most conserved parts of the HCV genome.
VIRAL REPLICATION AND LIFE CYCLE
Although peripheral blood mononuclear cells, B cells, T cells, and dendritic cells have been reported to support HCV replication, hepatocytes are the major site of viral replication.14,15 Understanding of the mechanisms of viral replication comes from studies in chimpanzees, extrapolation of the mechanisms used by other flaviviruses, and infection of cells in vitro with subgenomic replicons (segments of HCV RNA that are capable of amplification and synthesis of viral proteins but that do not produce mature viruses).
Early events in viral binding to the hepatocyte surface are still not completely understood (Fig. 79-1). HCV entry involves the attachment of envelope proteins E1 and E2 to cell surface molecules. The expression and function of CD81, a member of the tetraspan superfamily, are essential for HCV entry into hepatocytes.16 In addition, human scavenger receptor class B type 1 (SR-B1), a selective importer of cholesteryl esters from high-density lipoproteins (HDL) into cells, has been shown to interact with E2 and is essential for HCV entry.17 Whereas CD81 and SR-B1 are required early in the process of viral entry, claudin-1 (CLDN1), a tight junction component that is highly expressed on hepatocytes, is required later in the cell entry process.18 Heparin sulfated proteoglycans have also been shown to be essential for HCV cell entry.19 Other receptors are also likely required for viral entry.18 There is some evidence that the low-density lipoprotein (LDL) receptor is involved during endocytosis of HCV.20
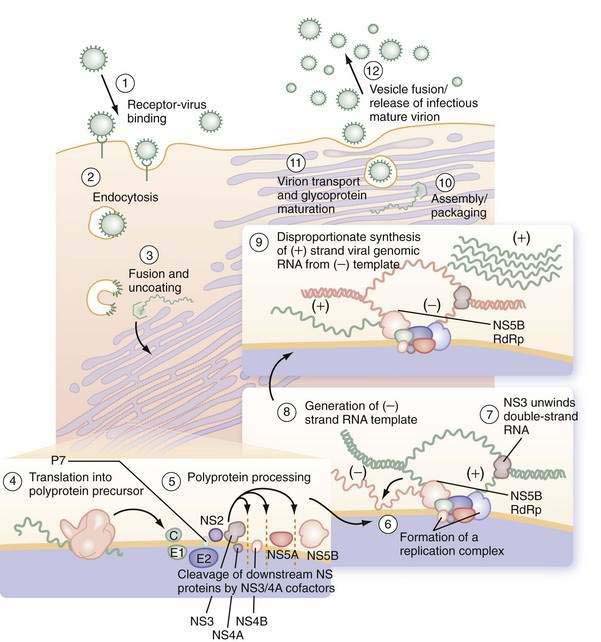
Figure 79-1. Putative life cycle of hepatitis C virus.
(Reproduced with permission from Pawlotsky JM, Chevaliez S, McHutchison JG. The hepatitis C virus life cycle as a target for new antiviral therapies. Gastroenterology 2007; 132:1979-98.) (See text for details and Fig. 79-2 for functions of the hepatitis C virus proteins.) NS, nonstructural; RdRp, RNA-dependent RNA polymerase.
Not only do viral receptors on hepatocytes play a role in entry of HCV into the cell, but also other cofactors affect the efficiency of viral infectivity. C-type lectins DC-SIGN (dendritic cell–specific intercellular adhesion molecule-3-grabbing nonintegrin; CD209) and L-SIGN (DC-SIGNr, liver and lymph node specific; CD209L) are expressed on dendritic cells and liver sinusoidal endothelial cells, respectively.21 Both receptors bind E2 on the HCV virus and facilitate infection of adjacent hepatocytes by acting as a capture and delivery mechanism. In addition, HDL increases infectivity of hepatocytes ten-fold, although the mechanism is not understood.22
Once the HCV virus attaches to the cell, endocytosis of the bound virion is presumed to occur, as in other flaviviruses. A pH drop in the vesicle causes conformational changes in the glycoproteins that lead to fusion of the viral and cellular membranes23 and release of viral RNA into the cytoplasm. In the cytosol, the 5′ UTR contains several highly conserved and structured domains that function as an IRES, which directs the RNA to its docking site on the endoplasmic reticulum and mediates cap-independent internal initiation of HCV polyprotein translation by recruiting both cellular proteins, including eukaryotic initiation factors (eIF) 2 and 3, and viral proteins.24,25
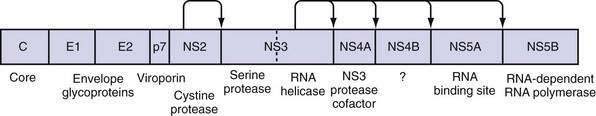
The large polyprotein generated by translation of the HCV genome is co- and post-translationally processed proteolytically into at least 11 viral proteins, including both structural (nucleocapsid [C], or p21; envelope 1 [E1], or gp31; and envelope 2 [E2], or gp70) and nonstructural (NS2, NS3, NS4A, NS4B, NS5A, and NS5B) proteins (Fig. 79-2).11,13 The functions of these specific proteins are described later in the chapter.
After polyprotein processing, NS4B expression causes the membrane alterations that are seen on electron microscopy as a membranous web.26,27 The replication complex associates viral proteins, cellular components, and nascent RNA strands and is essential for HCV replication, as demonstrated in replicon cell culture systems.28 HCV replication is catalyzed by the NS5B RNA-dependent RNA polymerase (RdRp). The positive-strand genomic RNA serves as a template for the synthesis of a negative-strand intermediate. Negative-strand RNA serves as a template for production of numerous strands of RNA of positive polarity that are used for polyprotein translation and synthesis of new intermediates of replication and are packaged into new virus particles.29
Finally, viral particle formation is initiated by the interaction of the core protein with genomic RNA in the endoplasmic reticulum, although the details of this process and subsequent export of mature virions from the hepatocyte are poorly understood.30,31 By analogy with pestiviruses, HCV packaging and release are likely to be inefficient because much of the virus remains in the cell. Following release, viral particles may infect adjacent hepatocytes or enter the circulation, where they are available for infection of another cell or host.
Virus Protein Function
The large polyprotein generated by translation of the HCV genome is cleaved by cellular and viral proteases to form structural and nonstructural proteins.11,13 The structural proteins are separated from the nonstructural proteins by the short membrane peptide p7, which is believed to be a viroporin, a protein that plays a role in viral particle maturation and release.32,33 At least one, and possibly three, alternative reading frame proteins (ARFP, or F for “frameshift”) exists.34–36 The exact number of alternative reading frames, the number of proteins that result, and the functions of the proteins are not known.36 One such protein is 17 kd in size; it can be expressed in vitro, and antibodies to it have been found in infected patients.
The core protein is first cleaved from the large polypeptide and then further processed by a host signal peptidase.11 In infectious HCV virions, core protein forms the viral nucleocapsid and binds RNA; it has many other functions as well. Core protein has been found attached to lipid rafts and the endoplasmic reticulum, and it translocates into the nucleus. When core protein attaches to lipid rafts, it recruits nonstructural proteins, resulting in the assembly of infectious virions. Core protein can also interact with the host immune system by inactivating the RNA silencing activity of Dicer, a cellular endoribonuclease that produces small interfering RNA to bind and target HCV RNA for destruction by the cell.37 Core protein can also bind to Janus kinase-1 (JAK1) and JAK2 and alter the activation of signal transducer activator of transcription (STAT) proteins, leading to impairment of interferon production.38 Extracellularly, core protein inhibits T-cell activation and proliferation, possibly by down-regulating co-stimulatory molecules on dendritic cells.39 Specific polymorphisms in core protein have also been associated with intracellular lipid accumulation40; this may be the result of facilitation of phosphorylation of insulin receptor substrate-1 (IRS-1), thereby leading to insulin resistance.41 Mutations in core protein have also been associated with an increased risk of hepatocellular carcinoma (HCC) in patients; core protein alone can cause HCC in transgenic mice.42
E1 and E2 proteins are cleaved from the polypeptide by host signal peptidase.11 The two proteins form highly glycosylated heterodimers and then tetramers that are essential for viral assembly (see earlier). They also mediate cell entry by binding to surface receptors.43 Subsequently, they are responsible for fusion between the host cell membrane and the viral envelope. Because E1 and E2 are expressed on the surface of the virion, they are targets of host antibodies. The first 27 amino acids of E2 form hypervariable region 1 (HVR1); alterations in HVR1 are believed to be an attempt by the virus at antibody-mediated immune evasion.
P7 is cleaved by the endoplasmic reticulum signal peptidase and forms an ion channel. This viroporin protein is essential for efficient assembly and release of infectious virions but not for cell entry.11,33 Because p7 is needed later in the viral life cycle, cleavage of the polypeptide is delayed.33
NS2 complexes with NS3 and zinc to form a cysteine protease, with two composite active sites, that autocatalytically cleaves NS2 from NS3.44 No other function of NS2 has been discovered to date. NS3 has several functions in addition to complexing with NS2 for autocatalytic cleavage of the NS2-NS3 site.44 Its function as a serine protease is markedly enhanced by its association with NS4A. The enzyme results in cleavage of the polyprotein at the NS3-NS4A, NS4A-NS4B, NS4B-NS5A, and NS5A-NS5B sites.11,45 The NS3 protease also cleaves and thereby destroys the function of Cardif and TRIF (Toll/interleukin receptor domain-containing adapter-inducing interferon-β), which are intermediates in two separate pathways of host-cell interferon secretion in response to viral infection.46–48 This property may have a significant effect in impairing the host response to HCV infection. Finally, a portion of the NS3 protein functions as a helicase that unwinds viral RNA as well as host DNA. The helicase function is dependent on adenosine triphosphate (ATP), may require dimerization of NS3, and progresses in discrete steps like an inchworm.12,49 NS4A complexes with NS3 and functions to stabilize the protease and helicase activities and anchor the complex to the endoplasmic reticulum membrane.11,12 It also regulates hyperphosphorylation of NS5A.50 The only known function of NS4B is to induce the formation of the “membranous web” on which HCV transcription occurs.26 NS5A binds zinc and forms homodimers that are bound to the endoplasmic reticulum membrane.12 NS5A is essential for viral replication and is believed to provide an RNA-binding site within the replication complex.51 In addition, NS5A inhibits apoptosis in infected cells,52,53 and some mutations confer improved sensitivity to interferon therapy.54 NS5B is the viral RNA-dependent RNA polymerase.11 The crystal structure elucidates the tunnel of the enzyme that directs single-stranded RNA into the active site.55 It can synthesize both negative-strand HCV RNA templates and positive-strand HCV RNA genomes.
GENOTYPES AND QUASISPECIES
HCV has an inherently high mutational rate that results in considerable heterogeneity throughout the genome.56 This high mutational rate is in part a consequence of the RNA-dependent RNA polymerase of HCV, which lacks 3′-to-5′-exonuclease proofreading ability that ordinarily would remove mismatched nucleotides incorporated during replication. An average of one error occurs for every 104 to 105 nucleotides copied. This phenomenon is favored by a high viral turnover rate; 1010 to 1012 virions are produced per day.57 A substantial proportion of newly synthesized viral genomes have alterations. Because of the functional differences in HCV proteins, genetic variation in some parts of the genome confers advantages by evading or inhibiting the host immune system, whereas other mutations may be lethal to the virus if they lead to defective replication machinery. Therefore, genetic variation is distributed irregularly along the genome. Each new genetic variant is produced in a single cell and may or may not spread through the liver and into the serum. The result is not only genetic diversity in the serum, but also compartmentalization of variant virions in different parts of the liver and perhaps in extrahepatic sites.
Because of the vast genetic variation, a classification scheme was devised whereby viral sequences are given a genotype and subtype. The first division used to describe the genetic heterogeneity of HCV is the viral genotype, which refers to genetically distinct groups of HCV isolates that have arisen during the evolution of the virus. Nucleotide sequencing has shown variation of up to 34% between genotypes.56 The most conserved region (5′ UTR) has a maximum nucleotide sequence divergence of 9% between genotypes, whereas the highly variable regions that encode the envelope proteins (E1 and E2) exhibit a nucleotide sequence divergence of 35% to 44% between genotypes. The sequences cluster into 6 major genotypes (designated by numbers), with sequence similarities of 60% to 70%, and more than 70 subtypes (designated by a lower case letter) within these major genotypes, with sequence similarities of 77% to 80%.56 In this scheme, the first variant, which was cloned by Choo and colleagues, is designated type 1a.58 The HCV genotype is an intrinsic characteristic of the infecting HCV strain and does not change over time; therefore, the genotype only needs to be determined once in an infected person. Mixed-genotype infections may be seen and reflect either coinfection with more than one HCV virus or methodologic problems in genotype testing. In addition, inter-genotypic HCV recombinants have been described59; these are thought to arise because of recombination between different genotypes in patients with repeated exposure. The recombination events have been reported to occur in or between NS2 and NS3.60
Global geographic differences exist in the distribution of HCV genotypes, as well as in the mode of acquisition. In the United States, genotype 1a is the most prevalent, accounting for approximately 57% of HCV infections, followed by genotype 1b in 17%, genotype 2 in 14%, genotype 3 in 7%, and genotype 4, 5, or 6 in less than 5%.61 Racial differences are seen in the prevalence of genotypes; approximately 90% of African Americans are infected with HCV genotype 1, whereas only 70% of whites and 71% of Hispanics are infected with genotype 1.62 In Europe, the most prevalent genotype is 1b (47%), followed by 1a (17%), 3 (16%), and 2 (13%).63 Genotype 4 is found mainly in Egypt, the Middle East, and Central Africa.64 In Egypt, approximately 15% of the population is infected with HCV, and more than 90% have HCV genotype 4. Because of the high prevalence rate in Egypt, genotype 4 represents 20% of the world’s HCV-infected population. Genotype 5, although originally isolated in South Africa, is also seen in specific regions of France, Belgium, and Spain.65 Genotype 6 is found predominantly in Asia. The distribution of genotypes is ever changing with immigration and alterations in the primary modes of viral transmission. Therefore, the frequencies of viral genotypes change over time.66
The second component of genetic heterogeneity is quasispecies generation.56 Quasispecies are closely related, yet heterogeneous, sequences of HCV RNA within a single infected person that result from mutations that occur during viral replication. The rate of nucleotide changes varies significantly among the different regions of the viral genome. The highest proportion of mutations is found in the E1 and E2 regions, particularly in HVR1. Even though this region represents only a minor part of the E2 region, it accounts for approximately 50% of the nucleotide changes and 60% of the amino acid substitutions within the envelope region.
The development of quasispecies may be one mechanism by which the virus escapes the host’s immune response and establishes persistent infection.67 During acute infection or during treatment, the lack of quasispecies is associated with viral clearance, and the development of numerous quasispecies is associated with viral persistence.68 In acute disease, patients in whom genetic variation in the HVR1 region develops after antibody seroconversion progress to chronic disease, whereas those in whom such genetic variation does not develop are more likely to achieve viral clearance.67 Genetic variation before seroconversion does not correlate with outcome, indicating that quasispecies formation results from antibody-mediated immune pressure. Interestingly, no intrinsically interferon-resistant variants of HCV have been defined, indicating that both viral and host factors play important roles in determining whether the virus persists or is cleared. An increased number of quasispecies has also been associated with more rapid progression to cirrhosis and the development of HCC.69
EPIDEMIOLOGY
INCIDENCE AND PREVALENCE
The worldwide seroprevalence of HCV infection, based on detection of antibody to HCV (anti-HCV), is estimated to be 3%, with more than 170 million people infected chronically. Marked geographic variation exists, with infection rates ranging from 1.3% to 1.6% in the United States to 15% in Egypt.1,64 Currently, between 3.2 and 5 million persons are infected with HCV in the United States.1 The prevalence rate is higher in persons 40 to 49 years old than in older or younger persons, in males (2.1%) than in females (1.1%), and in African Americans (3%) than in whites (1.5%).1 Other risk factors for HCV infection and the associated frequencies of infection are injection drug use (57.5%), blood transfusion before 1992 (5.8%), greater than 50 lifetime sexual partners (12%), and family income below the poverty level (3.2%). The current prevalence of HCV infection in the United States may be underestimated because the National Health and Nutrition Examination Survey (NHANES) data did not evaluate persons who are homeless, incarcerated, or in the military. Among incarcerated persons, 12% to 35% are positive for HCV RNA serum,70 whereas those in military service have a seroprevalence rate for anti-HCV of 0.5%.71
Worldwide, three different epidemiologic patterns of HCV infection have emerged: (1) previous exposure through health care with a peak prevalence in older persons; (2) exposure through intravenous drug use, the major risk factor since data first became available in about 1960, with a peak prevalence among middle-aged persons; and (3) ongoing high levels of infection in areas where high rates of infection occur in all age groups.63
Given the factors that influence viral diversity (see earlier), estimating the site of origin and age of HCV by phylogenetic analysis is difficult. The best estimate is that HCV originated in western and sub-Saharan Africa.72 Subsequent global spread probably occurred coincident with trade and human migration. Evolution of the virus led to a geographic distribution of genotypes, so that genotypes 1, 2, and 3 are most common in North America and Europe, genotype 4 is most common in the Middle East, and genotypes 5 and 6 are most common in Southeast Asia. In Japan, HCV transmission transitioned from constant to exponential growth in the 1920s, and the prevalence of HCV infection is highest in older persons.73 In Japan, and later in southern and Eastern Europe, health care–related procedures—particularly reuse of contaminated syringes—played a major role in viral spread. In the United States, Australia, and other developed countries, peak prevalence is in persons ages 40 to 49 years, and analysis of risk factors suggests that most HCV transmission occurred between the mid-1980s and the mid-1990s, through intravenous drug use. In Egypt, the spread of HCV increased exponentially from the 1930s to the 1980s because of mass vaccination campaigns with reuse of medical equipment.64 In Egypt and other developing countries, high rates of infection are observed in all age groups, suggesting that an ongoing risk of HCV acquisition exists.
In the United States, the incidence of acute hepatitis C is falling. The peak incidence was estimated to be 180,000 cases per year in the mid-1980s, but the rate declined to approximately 19,000 new cases by 2006.74 Many factors have contributed to the falling incidence of acute hepatitis C. In the 1980s, when blood was purchased from donors, 2% to 10% of blood units were infected with HCV, leading to a high rate of transfusion-acquired HCV infection.75 The institution of volunteer blood donation, creation of recombinant clotting factors, and implementation of HCV blood testing (between 1990 and 1992) dramatically decreased transfusion-acquired HCV infection.66
TRANSMISSION
Percutaneous Transmission
Blood transfusion, before the introduction of screening, and injection drug use are the most clearly documented risk factors for HCV infection. Following the introduction of anti-HCV screening of blood donors between 1990 and 1992, the number of transfusion-related cases of HCV infection declined sharply, and currently less than 1 case occurs per 2,000,000 units transfused.76,77
Injection drug use has always been the major route of HCV acquisition in the United States and accounts for an increasingly large portion of cases, at least 68% of new cases of HCV infection, since the virus was essentially eliminated from the blood supply.74 The prevalence of HCV infection in injection drug users ranges from 57% to 90%.1,78 Although risk factors for hepatitis B virus (HBV) and human immunodeficiency virus (HIV) infection overlap with those for HCV infection, the prevalence of HCV infection in this population is the highest among the three viruses. The majority of injection drug users become anti-HCV positive within six months of initiating injection drug use with shared paraphernalia.
Chronic hemodialysis is also associated with increased rates of HCV infection. The frequency of anti-HCV in patients on hemodialysis ranges from 11.6% in the United States to 55% to 85% in Jordan, Saudi Arabia, and Iran.79 Serologic assays for anti-HCV may underestimate the frequency of HCV infection in this relatively immunocompromised population, and virologic assays may be necessary for accurate diagnosis.80
Transmission may occur from infected patients to health care workers. A serologic survey of emergency department patients found that 18% were infected with HCV.81 The proportion with HCV infection was even higher in patients with a history of intravenous drug use (83%), past blood transfusion (21%), or a male homosexual lifestyle (21%). Although all potential routes of transmission of HCV infection to hospital workers are not obvious, needlestick injuries probably account for a large proportion of cases. Anti-HCV seroconversion rates are approximately 0.3% to 4% in longitudinal studies of health care workers after percutaneous inoculation from anti–HCV-positive sources, although the risk is dependent on the type of needle (hollow versus solid, infusion versus withdrawal), volume of inoculum, depth of injury, time the body fluid has spent ex vivo, level of viremia (viral load), and HIV status of the inoculating body fluid.82,83 In one study, 99% of surgeons in training experienced at least one needlestick by their final year of residency. Fifty-three percent of these injuries involved a high-risk patient, and only 49% were reported to the employee health service.84 As a result, as many as 16,000 new cases of HCV are estimated to have been transmitted to health care workers worldwide in 2000.85 Although less common, transmission of HCV also may occur from health care workers to patients. Because acute HCV infection often is subclinical, nosocomial transmission may occur with greater frequency than has been recognized previously. Strict adherence to universal precautions to protect health care workers and patients is critically important. At this time, no treatment is effective for post-exposure prophylaxis, and no data support such treatment even if it were available.
Nonpercutaneous Transmission
Nonpercutaneous modes of HCV transmission include sexual practices and childbirth. Available evidence indicates that transmission by nonpercutaneous routes occurs but is inefficient. From 10% to 20% of patients with HCV infection report that their only risk factor is sexual exposure to a partner with HCV infection. Most seroepidemiologic studies, however, have demonstrated anti-HCV in only a small proportion of sexual contacts of infected persons. In a large prospective study of monogamous seronegative partners of HCV-infected patients who denied anal intercourse and intercourse during menstruation, no instances of HCV transmission of the same sequenced virus occurred over a 10-year period of time.86 Therefore, many of the cases presumed to be the result of sexual transmission are likely the result of other, perhaps unreported or unrecognized, exposures. If the index sexual partner is infected with HIV or the partners engage in high-risk sexual practices, such as anal intercourse, however, the transmissibility of HCV is likely increased.87 Furthermore, epidemiologic studies have shown that persons with multiple sex partners have a higher prevalence of HCV infection.1 Whether sexually transmitted diseases promote transmission of HCV through breakdown of mucosal or immune barriers is unclear.
HCV-infected persons commonly are counseled to notify sexual partners of their HCV status. The risk of sexual transmission is negligible in monogamous couples that do not engage in high-risk sexual practices.86 Barrier methods should be recommended, however, to persons in non-monogamous relationships or those engaging in high-risk sexual practices.
Compared with the high efficiency of perinatal transmission of HBV infection (see Chapter 78), the risk of perinatal transmission of HCV infection is low, averaging 5.1% to 6.7% for HCV-monoinfected patients and two to three times higher for HIV-HCV-coinfected patients.88,89 Mothers with a high viral load are more likely to transmit HCV to their infants, a finding that may explain why infants born to mothers with HIV-HCV coinfection are at higher risk of HCV infection. Interestingly, the use of highly active antiretroviral therapy (HAART) in HIV-HCV-coinfected mothers may decrease the risk of perinatal transmission of both HIV and HCV.89 Data regarding the risk associated with vaginal delivery as opposed to cesarean delivery are uncontrolled, but evidence for a higher risk of HCV transmission with vaginal delivery is unconvincing. This issue remains controversial, and some authorities recommend elective cesarean section before membrane rupture.88
Although little data exist, the risk of HCV transmission from breastfeeding is negligible to small. The Centers for Disease Control and Prevention have concluded that breastfeeding by HCV-infected mothers is generally safe. Some authorities have suggested, however, that mothers with a high viral load (greater than 108 copies/mL, see later) may pose a risk.90 Because anti-HCV can be acquired passively by the infant, molecular testing for HCV RNA is required if the diagnosis of HCV infection is suspected. Infants of infected mothers should not undergo serologic testing for anti-HCV before the age of 18 months because maternal antibodies may persist in the infant’s serum and lead to diagnostic confusion.
Sporadic HCV Infection
The source of transmission is unknown in 9% to 27% of cases of HCV infection.78 Such sporadic HCV infection probably results from an undisclosed or unrecognized percutaneous route of infection. This presumption is supported by the observation that intranasal cocaine use is not considered a risk factor for HCV transmission (although it was considered a risk factor in the past).91 HCV infection can be acquired from non-commercial tattooing and body piercing when equipment is reused, shared, or improperly sterilized. Commercial tattooing is now well controlled and probably conveys little risk of HCV infection. Iatrogenic transmission of HCV is well documented in a variety of circumstances, most notably via contaminated multi-use vials and inadequately sterilized multi-use instruments and syringes, as seen with schistosomal treatment campaigns in Egypt.92
PATHOGENESIS
Determinants of persistence of HCV include (1) the evasion of immune responses through several viral mechanisms, (2) inadequate induction of the innate immune response, (3) insufficient induction or maintenance of an adaptive immune response, (4) the production of viral quasispecies, and (5) the induction of immunologic tolerance.67,93,94 In 55% to 85% of acute HCV infections, the net result of the host-virus interplay is the inability to clear virus despite the development of antibodies against several viral proteins. In the minority of patients in whom acute HCV resolves, an early and multispecific CD4+ T-cell proliferative response occurs, with predominance of type 1 CD4+ helper T (Th1) cells in the peripheral blood,95 most of which produce interferon-α. This “protective” response is still detected 18 to 20 years after infection in a majority of asymptomatic recovered patients but in only a minority of patients in whom chronic HCV infection develops.96 Although the immune response is essential in preventing viral persistence after acute HCV infection in 15% to 45% of cases, in those without viral clearance, the immune response mediates hepatic cell destruction and fibrosis.
VIRAL MECHANISMS
In chronically infected patients, the pathogenesis of liver damage is largely immune mediated. In a small subset of immunocompromised HCV-infected patients among both HIV-infected patients and organ transplant recipients, however, a syndrome termed fibrosing cholestatic hepatitis develops.97,98 Such cases are thought to result from direct viral hepatotoxicity of infected cells because viral levels are typically greater than 30 million copies/mL and hepatocytes contain enormous concentrations of virus and viral proteins.99 Survival in such patients is quite poor.
IMMUNE-MEDIATED MECHANISMS
HCV infection elicits an immune response in the host that involves both an initial innate response as well as a subsequent adaptive response. The innate response is the first line of defense against the virus and includes several arms such as natural killer (NK) cell activation and cellular antiviral mechanisms triggered by pathogen-associated molecular patterns (PAMPs) recognized by the cell.100–102 These processes can lead to apoptosis of infected cells within the first few hours of infection. NK cells, as the effector cells of the innate immune system, also produce tumor necrosis factor (TNF)-β and interferon-α, cytokines that are critical for dendritic cell maturation and subsequent induction of adaptive immunity.94 After this, however, the virus initiates a number of mechanisms that undermine the ability of the host to control the infection.
Virus-related disruption of the innate, and later adaptive, immune response occurs at several levels.93 NK cell function is slowed possibly because NK cell–mediated cytotoxicity and production of cytokines are interrupted when the HCV E2 protein binds its cellular receptor CD81.103,104 PAMPs activate several cellular processes including the JAK-STAT pathway and Toll-like receptor-3 (TLR-3), activation of both of which ultimately results in production of cellular interferons and interferon-regulated factors that convey antiviral properties to the cell.93 NS3/4 protease degrades TRIF, an essential intermediate in this pathway, and cleaves interferon promoter stimulator-1 (IPS-1), an intermediate in the signaling cascade, to activate interferon when retinoic inducible gene-1 (RIG-1) binds viral intermediates.47,105,106 In addition, HCV core protein promotes STAT-1 degradation, inhibits STAT-1 phosphorylation, promotes suppressor of cytokine signaling (SOCS) induction (an inhibitor of JAK-STAT signaling), and impairs interferon-stimulated gene factor-3 (ISGF3), a heterotrimer of STAT-1, STAT-2, and interferon-β promoter stimulator (IRF-9) from binding to the promoter regions of interferon-stimulated response elements (ISRE), thereby inhibiting transcription of interferon-response genes. Even when interferon-response genes are activated, NS5A and E2 both can disrupt protein kinase R (PKR) function to suppress translation, thereby allowing viral replication to continue.93 In addition, NS5A inhibits 2′-5′-oligoadenylate synthetase (OAS). 2′-5′-OAS is expressed in response to HCV infection and leads to HCV RNA degradation.93 Taken together, HCV is able to disorient the innate immune response at several levels, and these strategies appear to be pivotal in establishing the chronicity of infection.
The ability of HCV to impair the innate immune response prevents development of a vigorous adaptive immune response to the infection. NK cells do not adequately activate dendritic cells, and as a result, the priming of CD8+ and CD4+ T cells in HCV-infected patients is inadequate.107–109 Dendritic cells from HCV-infected patients are more likely to produce IL-10, a cytokine that inhibits antigen-specific T-cell responses. In addition, CD4+ T cells primed from dendritic cells of HCV-infected patients are more likely to produce IL-10. Even if an adequate T-cell response is created, HCV-infected patients have a large number of regulatory T cells in their portal tracts110; intrahepatic immune regulation by these cells has not been demonstrated but is presumed.
HCV-specific T cells are enriched at the site of viral replication, with an increased number in the liver when compared with the peripheral blood.95,111 CD8+ lymphocytes predominate, suggesting that cytotoxic T lymphocytes are the main perpetrators of hepatocellular injury. The T-cell immune response in the liver may result in direct lysis of infected cells and inhibition of viral replication by secreted antiviral cytokines.95,111
Whereas the cellular immune response plays a pivotal role in the pathogenesis of HCV infection, the importance of the humoral immune response is less clear. Antibodies to viral proteins are produced in low levels and do not appear to correlate with the stage of infection or immune reactivity. Furthermore, administration of high-titer HCV-enriched or HCV-specific immunoglobulin has little effect on viral levels or persistence in humans.112
CLINICAL FEATURES
ACUTE AND CHRONIC HEPATITIS C
HCV accounted for an estimated 20% of cases of acute hepatitis in 2006.74 Acute hepatitis C is rarely seen in clinical practice because nearly all cases are asymptomatic.87,113 Jaundice probably occurs in about 10% of patients with acute HCV infection, whereas 20% to 30% of patients present with nonspecific symptoms such as fatigue, nausea, and vomiting. HCV RNA is detectable within 2 to 3 weeks of exposure, and anti-HCV seroconversion occurs between day 15 and month 3. Serum aminotransferase levels peak at about the first month after exposure (Fig. 79-3), exceed 1000 IU/L in 20% of cases, and generally follow a fluctuating pattern for the first few months. In patients in whom jaundice develops, peak serum bilirubin levels usually are less than 12 mg/dL, and jaundice typically resolves within one month. Severe impairment of liver function and liver failure are rare. The presentation may be more apparent and the clinical course more severe when acute HCV infection occurs in patients who drink large amounts of alcohol or have coinfection with HBV or HIV.
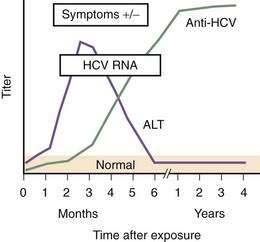
(Modified from the Centers for Disease Control and Prevention, www.cdc.gov/hepatitis/Resources/Professionals/Training/Serology/training.htm#one.)
The rate of viral persistence after acute infection varies, ranging from 45% to more than 90%. Age and gender clearly influence the risk of chronicity, with younger and female patients having the lowest rates of chronicity. Other factors that may play a role include the source of infection and size of inoculum (chronicity may be less common in injection drug users than in those who acquire HCV infection by blood transfusion), immune status of the host (chronicity rates are higher in persons with immunodeficiency states such as agammaglobulinemia and HIV infection), and the patient’s race (rates of viral persistence are higher in African Americans than in whites and Hispanic Americans in the United States). Finally, the rate of spontaneous clearance is higher in symptomatic patients in whom jaundice develops during acute infection than in those who remain asymptomatic.87,113,114 In patients with community-acquired hepatitis C in whom the infection resolves spontaneously, loss of HCV RNA from serum usually occurs within three to four months of the onset of clinical disease.114
Serum alanine aminotransferase (ALT) levels are usually elevated in patients with chronic HCV infection. Because levels commonly fluctuate, however, as many as one half of patients may have a normal ALT level at any given time.1 The ALT level may remain normal for prolonged periods of time in about 20% of cases,115 although transient elevations occur even in these cases.116 Persistently normal ALT levels are more common in women, and such cases typically are associated with lower serum HCV RNA levels and less inflammation and fibrosis on liver biopsy specimens.115
Most patients with chronic hepatitis C are asymptomatic before the onset of advanced hepatic fibrosis. Patients who have been diagnosed with chronic infection, however, often complain of fatigue or depression, and they consistently score lower than HCV-negative persons in all aspects of health-related quality of life (HRQL).117,118 Whether the decrease in HRQL is related to viral factors, social factors (e.g., intravenous drug use), social stigmatization, or worry related to the diagnosis itself is unclear. Nonetheless, HRQL scores improve if the patient achieves a sustained response to antiviral therapy (see later). Less common symptoms may include arthralgias, paresthesias, myalgias, sicca syndrome, nausea, anorexia, and difficulty with concentration. The severity of these symptoms may be, but is not necessarily, related to the severity of the underlying liver disease.
EXTRAHEPATIC MANIFESTATIONS
Patients with HCV infection may present with extrahepatic conditions, or these manifestations may occur in patients known to have chronic hepatitis C. Classification of the extrahepatic manifestations of HCV is shown in Table 79-1 and is based on the strength of available data to prove a correlation.119 Types 2 and 3 cryoglobulinemia, characterized by polyclonal immunoglobulin G (IgG) plus monoclonal IgM and polyclonal IgG plus polyclonal IgM, respectively, can both be caused by HCV infection. Among HCV-infected patients 19% to 50% have cryoglobulins in serum, but clinical manifestations of cryoglobulinemia are reported in only 5% to 10% of these patients and are more common in patients with cirrhosis. Symptoms and signs include fatigue, arthralgias, arthritis, purpura, Raynaud’s phenomenon, vasculitis, peripheral neuropathy, and nephropathy. The diagnosis is clear when a rheumatoid factor is detected, cryoglobulins are present, and complement levels are low in serum; however, the reliability of cryoglobulin measurements is dependent on proper handling and processing of the sample.
Table 79-1 Extrahepatic Manifestations of Hepatitis C Virus Infection
| Proven Associations |
| Autoimmune thyroiditis |
| B-cell non-Hodgkin’s lymphoma |
| Diabetes mellitus |
| Lichen planus |
| Mixed cryoglobulinemia |
| Monoclonal gammopathies |
| Porphyria cutanea tarda |
| Possible Associations |
| Chronic polyarthritis |
| Idiopathic pulmonary fibrosis |
| Non-cryoglobulinemic nephropathies |
| Sicca syndrome |
| Thyroid cancer |
| Renal cell carcinoma |
| Vitiligo |
Glomerular disease generally manifests as cryoglobulinemic nephropathy, membranoproliferative glomerulonephritis (MPGN), and membranous nephropathy.119,120 Cryoglobulinemic nephropathy manifests as hematuria, proteinuria, edema, and renal insufficiency of varying degrees, and on renal biopsy specimens it has features of MPGN. At diagnosis, 20% of patients with type 2 cryoglobulinemia have renal involvement, and renal involvement develops in another 35% to 60% over time. In about 15% of patients, cryoglobulinemic nephropathy progresses to end-stage kidney disease requiring dialysis.
Therapy should be considered in patients with symptomatic cryoglobulinemia. Cryoglobulinemia resolves in patients who achieve an SVR with pegylated interferon and ribavirin therapy (see later).119 Unfortunately, patients with significant renal involvement are at a disadvantage with respect to antiviral therapy because administration of ribavirin is generally contraindicated if the creatinine clearance is less than 50 mL/min. In these patients, treatment with rituximab should be considered.121,122 A durable clinical response to four doses of rituximab has been reported, although no prospective clinical trials have been completed to date. Prednisone, cyclophosphamide, other chemotherapeutic agents as well as plasmapheresis have been used with variable success; however, these approaches do not treat the underlying HCV infection.119 If cryoglobulinemia and renal disease improve with such treatment, then subsequent treatment of the HCV infection with pegylated interferon-α and ribavirin should be reconsidered.
HCV infection is associated with the development of B-cell non-Hodgkin’s lymphoma and monoclonal gammopathy of uncertain significance.119,123,124 The relative risk of lymphoma is small (1.28) in the United States.123 The most prevalent forms of lymphoma found in patients infected with HCV are follicular lymphoma, chronic lymphocytic lymphoma, lymphoplasmacytic lymphoma, and marginal zone lymphoma. In addition, marginal splenic lymphoma has been reported to regress after therapy for HCV infection alone. An estimated 8% to 10% of patients with type 2 cryoglobulinemia evolve into lymphoma over time. Despite the known association of HCV infection with lymphoma, HCV RNA does not integrate into the host genome and cannot be considered a typical oncogenic virus. Rather, HCV shows lymphotropism and may facilitate the development and selection of abnormal B-cell clones by chronic stimulation of the immune system. In addition, genetic rearrangements in B cells, specifically the Bcl2/JH rearrangement and the t(14;18) translocation, have been found in HCV-infected patients.125,126 Patients with B-cell genetic alterations are less likely to respond to antiviral therapy.127
Other extrahepatic manifestations of HCV infection include porphyria cutanea tarda, lichen planus, and sicca syndrome.119 In addition, insulin resistance and diabetes mellitus are associated with HCV infection. The SVR to antiviral therapy for HCV infection is reduced in insulin-resistant patients; however, if HCV can be eradicated, insulin resistance often improves, an observation that further supports the relationship between HCV infection and insulin resistance.128 Although associations between HCV infection and both thyroid cancer and idiopathic pulmonary fibrosis have been described, data about the effect of HCV eradication on disease progression are lacking (see Table 79-1). A myriad of other conditions have been observed in association with HCV infection, but a true link has not been firmly established for these disorders.119
Although not associated with disease, seropositivity for autoantibodies is found in many HCV-infected persons (e.g., antinuclear antibodies with a titer greater than 1 : 40 in 9%, smooth muscle antibodies with a titer greater than 1 : 40 in 20%, anti-liver-kidney microsomal antibodies in 6%).129 In addition, anti-thyroid peroxidase is found in serum in 5% to 12% of HCV-infected patients, although associated thyroid disease is found in only 2% to 5% of patients.119 Therefore, the diagnosis of an autoimmune condition in a patient with HCV infection can never be based on serology alone.
DIAGNOSIS
Several immunologic and molecular assays are used to detect and monitor HCV infection.130 The presence of anti-HCV in high titer in serum (generally an enzyme immunoassay [EIA] ratio greater than 9) indicates exposure to the virus but does not differentiate among acute, chronic, and resolved infection. Anti-HCV usually persists for many years, and perhaps for life, in patients after spontaneous resolution of infection or an SVR following antiviral therapy. Serologic assays are used initially for diagnosis, whereas virologic assays are required for confirming infection, monitoring response to treatment, and evaluating immunocompromised patients.130,131
INDIRECT ASSAYS
EIAs detect antibodies against different HCV antigens. The time course of the development of symptoms, detection of anti-HCV, and appearance of HCV RNA after acute infection is shown in Figure 79-3. Three generations of EIAs have been developed. The latest, third-generation, EIAs detect antibodies against HCV core, NS3, NS4, and NS5 antigens as early as 7 to 8 weeks after infection, with sensitivity and specificity rates of 99%.131 Despite ongoing viral replication, serologic test results can be negative in patients who are on hemodialysis or are immunocompromised.80 Because the performance characteristics of third-generation EIAs are so good, confirmation with a recombinant immunoblot assay (RIBA) is no longer required. Instead, patients who are anti-HCV positive should undergo HCV RNA testing to determine if they have active viremia or have cleared the infection.
DIRECT ASSAYS
Two general types of direct assays exist: qualitative and quantitative HCV RNA tests.130 Qualitative HCV RNA nucleic acid tests (NAT) only report whether HCV RNA is found in serum or not and do not quantitate the amount of HCV RNA. These tests should be used only for screening purposes now (e.g., screening of blood donated to a blood bank) and should not be used in clinical practice. As a result of NAT technology, transfusion-related HCV infection has decreased to less than one case per two million units of blood transfused.76,77
Unlike NAT testing, quantitative HCV RNA tests are essential for monitoring the response to antiviral therapy (see later). Currently “real-time” tests, such as Taqman, are performed using polymerase chain reaction (PCR) methodology, with a lower limit of detection of 10 to 15 international units (IU)/mL.132 These assays have a linear dynamic range of 1 to 7 log10 IU/mL and are the preferred testing method in practice. Transcription-mediated amplification (TMA) is also extremely sensitive, but available assays are not quantitative in the lower dynamic range of the test. The advantages of these very sensitive tests include positivity within one to three weeks after acute infection and detection of low-level residual infection during antiviral therapy.
HCV Genotyping
Identifying the genotype of HCV can be accomplished by several methods. The most accurate method is PCR and direct sequencing of the NS5B or E1 region; however, this approach is not practical in clinical practice. HCV genotyping can be done by evaluating type-specific antibodies and has a 90% concordance in immunocompetent patients when results are compared with sequence analysis of the HCV genome.131 Testing can also be accomplished with reverse hybridization to genotype-specific probes, restriction fragment length polymorphism analysis, or PCR amplification of the 5′ noncoding region of the HCV genome. These tests have 92% to 96% concordance with the correct genotype; genotype 1 is identified with the highest accuracy. Because of mutations in the regions studied, regardless of the technique used, errors in subtype identification occur in 10% to 25% of cases. A line-probe assay (Inno-LIPA) using genotype-specific probes for reverse transcription of the 5′ portion of the HCV genome is currently the most popular commercial assay for HCV genotyping.133
SELECTION OF SEROLOGIC AND VIROLOGIC TESTS
Screening in Blood Banks
The risk of acquiring HCV infection from blood products has declined dramatically since blood donors have been screened routinely. Many blood banks have now switched from third-generation EIA testing to NAT testing, and this change has decreased the risk of transfusion-acquired HCV infection to one infection per two million units of blood transfused.76,77
Diagnosis Following Known Exposure
Following an occupational or recreational exposure or in the context of mother-to-infant transmission, the diagnosis of HCV infection is now based on HCV RNA testing by the most sensitive molecular method, usually a real-time PCR assay. If transmission has occurred, HCV RNA is detectable in serum 1 to 3 weeks following exposure, whereas anti-HCV may not be seen for 7 to 8 weeks following exposure. Treatment should be considered in adults with acute exposure because the risk of chronicity is high without treatment and clearance of HCV RNA with antiviral therapy is likely (see later). Therefore, early diagnosis of HCV infection is important in these settings.130 In patients with a continued risk of infection, as through injection drug use, periodic testing for HCV RNA should be done. Because spontaneous clearance of HCV RNA from serum is more likely in infants than in adults, an infant should not be tested until 18 months after birth if the mother is HCV RNA positive.
LIVER BIOPSY AND NONINVASIVE ASSESSMENT OF FIBROSIS
The risk of progressive hepatic injury from HCV infection varies considerably, with some patients showing little or no progression after decades of infection and others progressing rapidly to cirrhosis.134 Therefore, an assessment of the degree of liver injury is usually advisable. This assessment is usually done by percutaneous liver biopsy (Table 79-2), but indirect and noninvasive methods to assess liver injury and fibrosis are under study and becoming commercially available.
Table 79-2 Reasons to Perform a Liver Biopsy in a Patient with Hepatitis C
| Assessment of the need for surveillance for hepatocellular carcinoma |
| Evaluation for concomitant liver diseases |
| Guidance for decisions regarding treatment of hepatitis C |
| Staging of fibrosis |
Examination of liver biopsy specimens is used to quantify hepatic injury into discrete grades of inflammation and stages of fibrosis.134,135 Several scoring systems have been used and differ in range of possible scores and definitions (Fig. 79-4). The first system used was the Histology Activity Index (HAI) described by Knodell and colleagues. The components of this system include periportal inflammation and necrosis (graded as 0 to 10), lobular inflammation and necrosis (0 to 4), portal inflammation (0 to 4), and fibrosis (0 to 4). This scoring system combines inflammation and fibrosis into one score. Scheuer created a simplified scoring system that separates grade from stage: Portal inflammation and interface hepatitis (0 to 4), lobular activity (0 to 4), and fibrosis stage (0 to 4). The Ishak system is a modification of Knodell’s system but separates histologic grade from stage. Ishak’s fibrosis scores range from 0 to 6 (1 or 2, portal fibrotic expansion; 3 or 4, bridging fibrosis; 5 or 6, cirrhosis) (Fig. 79-5). The higher number of gradations of fibrosis has made the Ishak system popular for scoring progression of fibrosis in clinical trials. Currently, the METAVIR scoring system is the most popular; it is simpler than all the aforementioned systems. Inflammation is graded from 0 to 4 (none, mild, moderate, and severe), and fibrosis is staged from 0 to 4 (1, portal fibrotic expansion; 2, portal fibrosis with septa formation; 3, bridging fibrosis; 4, cirrhosis).
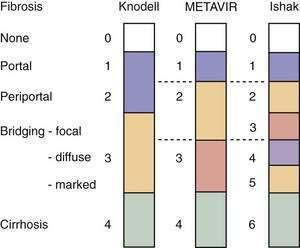
Figure 79-4. Comparison of the Knodell, METAVIR, and Ishak hepatic fibrosis staging systems. The METAVIR staging system is similar to the Scheuer system. Portal, periportal, bridging, and cirrhosis describe the degree (stage) of fibrosis (see also Fig. 79-5).
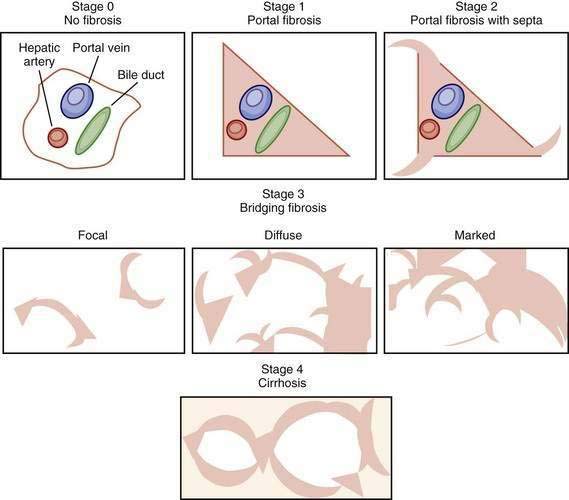
Figure 79-5. Visual depictions of fibrosis staging (METAVIR system) in patients with chronic hepatitis C. Stage 0 represents the absence of abnormal fibrosis, stage 1 shows portal fibrosis, and stage 2 shows portal fibrosis with septa. Bridging fibrosis (stage 3) is a continuous process ranging from focal periportal fibrosis with occasional bridging between portal structures to marked bridging without nodule formations, sometimes called early cirrhosis. Depending on the staging system (see Figure 79-4
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


