Chapter 82 Hepatic tumors in childhood
Overview
An appreciation of hepatic segmental anatomy (see Chapter 1A, Chapter 1B ) has led to major advances in hepatic surgery, especially for tumors. In addition, the irresistible but still mysterious stimulus to hepatic regeneration has allowed larger and more extensive resections. In small infants, 85% of the liver can be removed safely, greatly increasing the scope for cure. Advances have also been made in understanding tumor biology and clinical behavior. This chapter addresses benign and malignant tumors of the liver and biliary tract encountered in infancy, childhood, and adolescence.
History
Between 310 and 280 bce, Herophilus and Erasistratus first presented a description of hepatic anatomy. In the late 1880s, hepatic resection was attempted, but advances in anesthesia and antisepsis would be required before a successful outcome could be realized. Wendel used avascular anatomic planes in the liver to perform a hepatic resection in 1910 (McClusky et al, 1997), and progress in hepatic surgery has been based on an appreciation of hepatic segmental anatomy as described by Couinaud (Bismuth, 1982; Couinaud, 1986, 1992; see also the Introduction to this text). The distribution of the portal and hepatic veins delimits each hepatic segment, which has a unique portal vein and hepatic artery branch and bile duct. Knowledge of this anatomy allows control of the vascular structures before division of the hepatic parenchyma, making major hepatic resections feasible (see Chapter 1A, Chapter 1B ).
Bloodless hepatic dissection is crucial in infants and small children, who may have a total blood volume of less than 1 L. In the pediatric literature, Martin and Woodman (1969) reported that hepatoblastomas could be treated by hepatic lobectomy, and modern hepatic resection is soundly based on principles of segmental hepatic anatomy.
A second important historic finding was the sensitivity of some tumors, especially hepatoblastoma, to systemic chemotherapy (Fegiz et al, 1977). Chemotherapy caused significant reductions in tumor volume, and previously unresectable hepatoblastomas became resectable (Filler et al, 1991; Reynolds, 1995). Presently, the standard of practice is to administer neoadjuvant systemic chemotherapy to patients with hepatoblastoma, unless the tumor is clearly resectable at diagnosis.
In addition, appreciation of the biology of hepatic epithelial malignancies has increased, especially the differences between hepatoblastoma and hepatocellular carcinoma (HCC). These differences include the relatively good prognosis of hepatoblastoma compared with HCC in childhood, the importance of complete surgical resection of the primary hepatic tumor, and the association of hepatoblastoma with certain clinical syndromes (Exelby et al, 1975; Koishi et al, 1996; Schneid et al, 1997; Simms et al, 1995; Tsai et al, 1996; Vaughan et al, 1995). Finally, the first application of hepatic transplantation to a childhood liver tumor was reported by Heimann and colleagues in 1987, and a series of pediatric liver tumor patients treated by hepatic transplantation was reported by Tagge and colleagues in 1992. There is continued interest in using this modality for unresectable hepatic malignancies in childhood and adolescence (Pichlmayr et al, 1995; Pinna et al, 1997; Superina & Bilik, 1996).
Malignant Tumors
Primary malignant liver tumors constitute approximately 1.1% of childhood malignancies in Western nations. The overall incidence of primary liver cancer, as published by the Surveillance Epidemiology and End Results (SEER) program, is 5 cases per 1 million children in the 0 to 4 year age group and 1 case per 1 million in the 5 to 9, 10 to 14, and 14 to 19 year age groups (National Cancer Institute, 1995). Liver cancers constitute 0.5% to 2% of all pediatric solid tumors and about 5% of abdominal tumors in childhood (Weinberg & Finegold, 1983). The distribution of the most common malignant hepatic tumors is depicted in Figure 82.1. Hepatoblastoma is the most common, and its treatment is a success story in pediatric oncology.
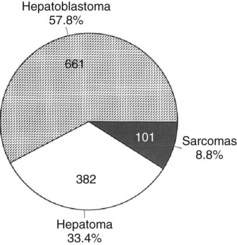
FIGURE 82.1 The frequency distribution of malignant hepatic tumors in childhood as compiled from reported large series.
(From Exelby PR, et al, 1975: Liver tumors in children in the particular reference to hepatoblastoma and hepatocellular carcinoma: American Academy of Pediatrics Surgical Section Survey 1974. J Pediatr Surg 10:329-337; and Weinberg AG, Finegold MJ, 1983: Primary hepatic tumors of childhood. Hum Pathol 14:512-537.)
Hepatoblastoma
Incidence
Hepatoblastomas are the most common primary hepatic tumors of childhood, constituting 43% to 64% of all hepatic neoplasms in one large series (Mann et al, 1990; Stocker, 1994; Weinberg & Finegold, 1983). Hepatoblastoma accounts for 91% of primary hepatic tumors in children younger than 5 years (Darbari et al, 2003) but comprises less than 1% of hepatic malignancies when adult age groups are included (Kaczynski et al, 1996). The Liver Cancer Study Group (LSCG) of Japan (1987) identified 30 hepatoblastomas (0.6%) in a cohort of 4,658 patients of all ages diagnosed over a 2-year period.
Hepatoblastoma affects 1 child younger than 15 years old in 1 million per year (Finegold, 1994), and approximately 50 to 70 new cases per year are reported in the United States, with a male/female ratio of 1.7 : 1 (Lampkin et al, 1985). Although hepatoblastoma has been reported sporadically in adults (Bortolasi et al, 1996; Harada et al, 1995; Inoue et al, 1995; Kacker et al, 1995; Parada et al, 1997), the median age at diagnosis is about 18 months, and most cases occur before the age of 3 years (Exelby et al, 1975). Hepatoblastoma is the most prevalent malignant neoplasm of the fetus and neonate and results in death within 2 years if not treated (Dehner, 1978; DeMaioribus 1990; Isaacs, 1985, 2007; Patterson et al, 1985). Finally, the incidence of hepatoblastoma may be increasing (Blair & Birch, 1994). The incidence of hepatoblastoma was 0.6 per 1 million in the years 1973 through 1977, and it increased to 1.2 per 1 million from 1993 through 1997 (Darbari et al, 2003).
Hepatoblastoma may occur in siblings (Fraumeni et al, 1969; Ito et al, 1987; Napoli & Campbell, 1977; Surendran et al, 1989). It is most strongly associated with familial polyposis (Giardiello et al, 1996; Iwama & Mishima, 1994), Gardner syndrome (Hartley et al, 1990), and Beckwith-Wiedemann syndrome (Koishi et al, 1996; Tsai et al, 1996). In familial polyposis, incidence of hepatoblastoma seems to be increased in first-degree relatives of the patients with polyposis. Beckwith-Wiedemann syndrome is associated with Wilms tumor, rhabdomyosarcoma, adrenocortical carcinoma, and hepatoblastoma with a possible association between hepatoblastoma and trisomies 2, 8, 18, and 20 (Bove et al, 1996; Tomlinson, 2005).
Hepatoblastoma is also associated with low birth weight (Ikeda et al, 1997; Reynolds et al, 2004). It is unknown whether the causative agent is developmental abnormalities associated with prematurity or interventions, such as early total parenteral nutrition. These tumors also are reported in patients with congenital anomalies, such as cleft palate, and cardiovascular and renal anomalies, including multicystic kidney and absence of the right adrenal gland (Rao et al, 1989). There are also at least two reports of hepatoblastoma occurring in patients with biliary atresia (Taat et al, 2004). No evidence associates hepatoblastoma with hepatitis B or C infection or any other chronic viral hepatitis. These patients usually do not have cirrhosis or inborn errors of metabolism.
Pathology
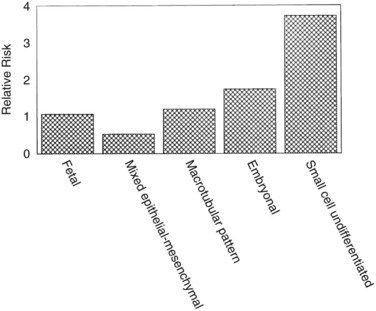
Hepatoblastomas are large tumors that can contain fibrous bands, producing a spoked-wheel appearance (Jha et al, 2009). The five histologic subtypes observed in hepatoblastoma are 1) fetal, 2) embryonal, 3) mixed epithelial, 4) mesenchymal/macrotubular, and 5) anaplastic or small-cell undifferentiated. These subtypes are differentiated based on the findings with light microscopy, but all tumor cells appear smaller than nonneoplastic hepatocytes. Extramedullary hematopoiesis is notably present and may be related to constitutive cytokine production by the tumor cells (von Schweinitz et al, 1995a). The fetal type grows in trabeculae and resembles fetal hepatic cells, whereas embryonic hepatoblastoma cells grow in incohesive sheets and resemble embryonic cells. Some hepatoblastomas contain mesenchymal tissue along with the epithelial component. Calcification also may appear in these tumors, and one patient was reported with osteosarcomatous elements in the hepatoblastoma and associated pulmonary metastases (Alcantar, 1985). The anaplastic or small-cell type consists of small, round blue cells reminiscent of neuroblastoma. This subtype is rare but particularly virulent, with a strong metastatic potential (Dehner & Manivel, 1988). The importance of subtyping in hepatoblastoma is due to the association between prognostic risk and subtype, illustrated in Figure 82.2 (Gonzalez-Crussi et al, 1982; Lack et al, 1982). Some studies have indicated that the fetal histologic subtype has a better prognosis; in contrast, patients with the rare anaplastic variant usually do poorly (Dehner & Manivel, 1988).
Basic Biology
Few cellular models of hepatoblastoma exist, and immortalized cell lines have been difficult to establish. One cell line, isolated from a human hepatoblastoma, clearly expresses the MYC and Hras1 oncogenes and epidermal growth factor receptor (EGFR) (Manchester et al, 1995). Antibodies that blocked the EGFR inhibited cell growth, but the significance of oncogene expression remains to be determined. A new cell line was established from primary hepatoblastoma tumor tissue and contains an identical genotype to tumor cells, with morphologic, molecular, and immunohistochemical confirmation.
In immunodeficient mice, hepatoblastoma is tumorigenic (Chen, 2009). Elevated hepatocyte growth factor levels have been demonstrated in the serum of 10 (45%) of 22 patients with hepatoblastoma (von Schweinitz et al, 1997a). Addition of hepatocyte growth factor causes proliferation in hepatoblastoma-derived cell lines.
Small epithelial cells, characteristic of hepatic stem cells, have been observed in human hepatoblastomas of various subtypes (Ruck & Xiao, 2002). Additionally, various genetic abnormalities have been reported in hepatoblastoma. Chromosome 8q amplification is associated with a worsened prognosis and has been correlated with overexpression of the transcription factor pleomorphic adenoma gene 1 (PLAG1) (Zatkova et al, 2004). Telomerase and its regulatory protein expression levels have been correlated with poor outcome in human hepatoblastoma (Hiyama et al, 2004), and tamoxifen may inhibit hepatoblastoma cells by reducing telomerase levels (Brandt et al, 2005).
Loss of heterozygosity on chromosome 11p15.5, the region associated with Beckwith-Wiedemann syndrome, and on chromosome 1p36 has been described in hepatoblastoma (Albrecht et al, 1994; Kraus et al, 1996). Investigations into both of these regions suggest that each may contain a tumor suppressor gene, but this has not been proven. Trisomy 20 and trisomy of all or part of chromosome 2 also have been reported (Swarts et al, 1996). In addition, an abnormality of chromosome 2q may provide a common genetic link between hepatoblastoma and rhabdomyosarcoma (Rodriguez et al, 1991). Finally, frequent genetic losses found using comparative genomic hybridization included regions 13q21-q22 (28%) and 9p22-pter (22%), and the most frequent genetic gains were on chromosomes 2q23-q23 (33%) and 1q24-q25 (28%; Gray et al, 2000). Recently, differentially expressed microRNA has been shown to be deregulated in hepatoblastoma (Magrelli et al, 2009), and mutations of β-catenin have also been recognized (Curia et al, 2008).
Perhaps the most exciting insight is the association between hepatoblastoma and familial adenomatous polyposis syndrome (Bala et al, 1997; Cetta et al, 1997). In one study of 13 hepatoblastomas obtained from nonfamilial adenomatous polyposis patients, 69% had mutations in the adenomatous polyposis coli (APC) gene (Oda et al, 1996). In one case of siblings with hepatoblastoma, a shared APC gene mutation was identified (Thomas et al, 2003). In addition, the well-known thrombocytosis associated with untreated hepatoblastoma is fascinating, as is the presence of extramedullary hematopoiesis in these tumors. It was shown that hepatoblastoma cells secrete interleukin (IL)-1β, which causes secretion of IL-6 from surrounding fibroblasts and endothelial cells (von Schweinitz et al, 1993). Other factors, such as erythropoietin and stem cell factor, have been localized to the cytoplasm of hepatoblastoma cells. Thrombopoietin has been identified in hepatoblastoma tissue and serum from a patient, but its correlation with the thrombocytosis associated with this neoplasm is unclear (Komura et al, 1998).
Clinical Findings
The most common presenting sign of hepatoblastoma is an asymptomatic abdominal mass. The child is often in good health, and the tumor usually is discovered incidentally, when an attentive parent, grandparent, or clinician discovers the mass on a routine examination or while bathing the child (Fabre et al, 2004). Patients with the anaplastic variant of hepatoblastoma, who often have distant metastases at diagnosis, are more frequently symptomatic. Accompanying symptoms such as pain, irritability, minor gastrointestinal disturbances, fever, and pallor occur in smaller numbers of patients. Significant weight loss is unusual, although patients may fail to thrive. In most series of hepatoblastomas and HCCs, a few patients present acutely with tumor rupture and intraperitoneal hemorrhage (Brown et al, 1993). Rarely, hepatoblastoma manifests with sexual precocity secondary to a β-human chorionic gonadotropin (hCG)-secreting tumor (Muraji et al, 1985), and one patient with a hepatoblastoma was reported presenting with a biliary fistula (Daniel & Kifle, 1989). Finally, hepatoblastoma may present as a cardiac tumor (Wang et al, 2003).
Measurement of serum α-fetoprotein (AFP) is well established as an initial tumor marker in the diagnosis of hepatoblastoma and a means of monitoring the therapeutic response (Van Tornout et al, 1993). The normal level in most laboratories is less than 20 ng/mL, whereas the AFP level at diagnosis in hepatoblastoma patients can range from normal to significantly elevated (7.7 × 106 ng/mL); it is estimated that the AFP is elevated in 84% to 91% of patients with hepatoblastoma (Lack et al, 1982). One study reported a mean AFP level in hepatoblastoma of 3 million ng/mL, whereas the mean in pediatric patients with HCC was about 200,000 ng/mL (Ortega et al, 1991). In infants younger than 1 year, the AFP is normally elevated and is highest at birth (Fig. 82.3).
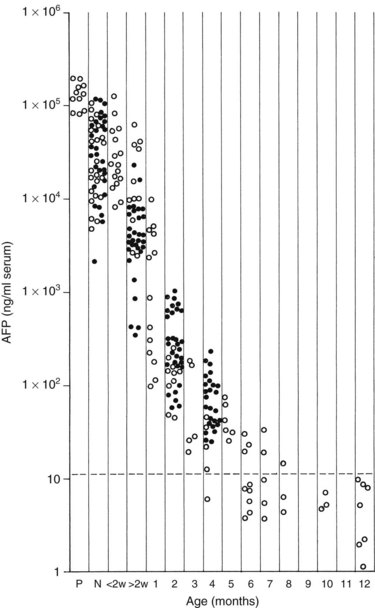
FIGURE 82.3 Graph shows the time decay of α-fetoprotein (AFP) levels in normal infants during the first year of life.
(Data from Wu JT, et al, 1981: Serum alpha fetoprotein [AFP] levels in normal infants. Pediatr Res 15:50-52.)
Some authors suggest that subfractionation more reliably indicates whether the increased AFP is secondary to a hepatoblastoma or HCC, an endodermal sinus tumor, or benign liver disease (Tsuchida et al, 1997). The half-life of AFP is about 6 days, and in one study, 24 (77%) of 31 patients had levels decline by at least 1 log before second-look surgery (Walhof et al, 1988). Of these patients, 16 (50%) of 32 eventually had AFP levels decline to normal at the end of therapy and had no clinical or radiographic evidence of hepatoblastoma at this point. Finally, 15 (94%) of 16 patients who attained a complete response also showed a decline in AFP levels of 2 logs or more before second-look surgery (Van Tornout et al, 1997). A large, early decline in AFP levels was an independent predictor of survival in multivariate analysis; it has also been stated that a low initial AFP level is associated with worse survival (von Schweinitz et al, 1995b), but this has not been confirmed in multivariate analysis (von Schweinitz et al, 1994a). Anaplastic hepatoblastomas may also be associated with lower AFP levels (Tsunoda et al, 1996).
When interpreting the AFP level, it is important to realize that normal levels are very high at birth and decrease over the first 6 months of life. Premature newborns may have AFP levels in the range of 100,000 ng/mL. Term newborns also can have relatively high levels (104 to 105 ng/mL). By 2 months of age, most infants have levels ranging from 100 to 1000 ng/mL, and by 6 months, levels should be less than 100 ng/mL. Usually, levels decrease to normal (<20 ng/mL) after 6 to 7 months, but levels may remain elevated for 1 year after birth (Ohama et al, 1997). AFP may also be elevated in the setting of liver damage, liver regeneration, or in the presence of another tumor.
Imaging
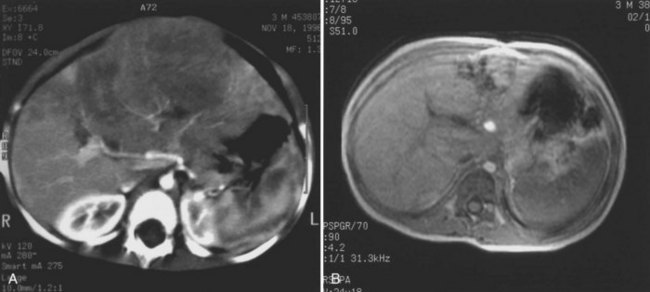
The first imaging study is usually an abdominal ultrasound (US) (see Chapter 13). If duplex technique is used, tumor vascularity can be gauged, and the hepatic veins can be assessed (Bates et al, 1990). The ultrasonographer also should search for anomalies of the genitourinary system and rule out tumor thrombus in either the vena cava or the hepatic veins. Computed tomography (CT) (see Chapter 16) is useful to identify pulmonary metastases, identify diffuse hepatic involvement, and determine resectability. Oral and intravenous contrast material is used (Fig. 82.4A). CT scans can be performed quickly and can be completed in less than 2 minutes in helical scanners; this greatly shortens the required period of sedation for infants or small children, and it has the added advantage of being a quick and reliable screening method for pulmonary metastases. Hepatoblastoma will appear as a well-demarcated tumor, without a capsule. CT angiography (CT portography) uses CT with fine cuts and an increased amount of intravenous contrast material to image hepatic tumors and the hepatic venous anatomy. CT portography may provide as much information as magnetic resonance imaging (MRI) (see Chapter 17), which is useful for evaluating hepatic lesions and their relationship to vascular structures. MRI can show the hepatic veins, vena cava, and bile ducts (Ohnuma et al, 1991). MRI of a hepatoblastoma patient after neoadjuvant chemotherapy is shown in Figure 82.4B. Positron emission tomography (PET) has been used to identify hepatoblastoma recurrence and to search for sites of metastatic disease, but it may not be reliable for lesions smaller than 6 to 10 mm (Wong et al, 2004).
Staging
In the United States, the commonly used staging system is that from the Children’s Oncology Group (COG), based on operative findings (Table 82.1). A tumor-node-metastasis (TNM) classification has also been used (Table 82.2; Brower et al, 1998). The PRE-Treatment EXTent (PRETEXT) system of disease staging has been used extensively by the International Society of Pediatric Oncology liver group (SIOPEL) (Fig. 82.5; Aronson et al, 2005). It relies on radiographic findings prior to any therapy, including surgery, and does not take into account the independent surgeon’s judgment at the time of surgery regarding resectability. This staging system is based on Couinaud’s system of segmentation of the liver and is thought to predict the degree of tumor infiltration, the extent of surgical resection, and the complexities involved in the resection (Couinaud, 1992; Otte, 2010). This system classifies the tumor into one of four categories, depending on which sections of the liver do not include tumor (Table 82.3; Roebuck et al, 2007). Additional criteria added in 2005 (Table 82.4) further classify these tumors (Otte, 2010; Roebuck, 2007).
Table 82.1 Children’s Oncology Group Staging for Hepatoblastoma
| Stage I | Complete Resection |
|---|---|
| Favorable histology | Purely fetal histology with a low mitotic index |
| Other histology | All other stage I tumors |
| Stage II | Gross total resection with microscopic residuals or total resection with preoperative or intraoperative rupture |
| Stage III | Unresectable tumors as determined by the attending surgeon, partially resected tumors with macroscopic residual involvement or any tumor with lymph node involvement |
| Stage IV | Measurable metastatic disease to lungs or other organs |
From Finegold MJ, et al, 2008: Liver tumors: pediatric population. Liver Transpl 14(11):1545-1556.
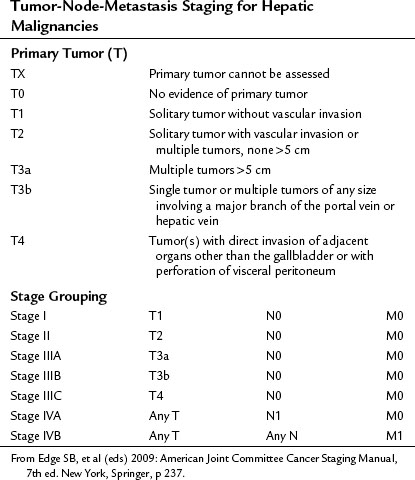
FIGURE 82.5 The PRE-Treatment EXTent (PRETEXT) staging system used by the International Society of Pediatric Oncology.
Table 82.3 PRETEXT Staging System
| PRETEXT Number | Definition |
|---|---|
| I | One section is involved, and three adjoining sections are free |
| II | One or two sections are involved, but two adjoining sections are free |
| III | Two or three sections are involved, and no two adjoining sections are free |
| IV | All four sections are involved |
PRETEXT, PRE-Treatment EXTent
From Roebuck DJ, et al, 2005: PRETEXT: a revised staging system for primary malignant liver tumours of childhood developed by the SIOPEL group. Pediatr Radiol 37:123-132.
Table 82.4 PRETEXT Staging: Additional Criteria
| C: Caudate lobe involvement | |
| E: Extrahepatic abdominal disease | |
| F: Tumor focality | |
| H: Tumor rupture or intraperitoneal hemorrhage | |
| M: Distant metastases | |
| N: Lymph node metastases | |
| P: Portal vein involvement | |
| V: Involvement of the IVC and/or HVs |
IVC, inferior vena cava; HVs, hepatic veins
Reprinted with permission from Otte JB, 2010: Progress in the surgical treatment of malignant liver tumors in children.
PRETEXT was compared with pathologic findings in 110 patients and was correct in 51%, overstaged in 37%, and understaged in 12%. The authors compared this system with the Children’s Cancer Group/Pediatric Oncology Group (CCG/POG) and TNM schemes and claimed a better correlation with risk status. This study analyzed data from patients who had neoadjuvant chemotherapy, whereas a recent study from the COG analyzed data from patients with a hepatoblastoma at diagnosis and reported that both the COG stage and PRETEXT were useful prognostic indicators. The PRETEXT system has been described as showing improved predictive value for survival compared with other staging classifications (Aronson et al, 2005). Moreover, this system can be valuable for recognizing patients who are surgical candidates (PRETEXT stages I and II) and those who may benefit from lower dose chemotherapy (Meyers et al, 2009). It is recommended that all liver tumor patients in future COG studies undergo PRETEXT staging.
Treatment
Multiple studies support the effectiveness of systemic chemotherapy combined with complete surgical resection of the primary hepatic tumor (Gauthier et al, 1986; von Schweinitz et al, 1994a, 1994b, 1995b). Survival depends on removal of the primary liver tumor, when imaging suggests that complete resection is feasible.
The first clinical decision is whether to initiate neoadjuvant chemotherapy or proceed with resection. Often, resection is not feasible if tumors are large and involve both hepatic lobes. Preoperative (neoadjuvant) chemotherapy results in tumor shrinkage and makes subsequent resection easier (Reynolds, 1995). In one study, the rate of shrinkage was high after initiation of chemotherapy, but it declined after two cycles had been administered (Fig. 82.6; Medary et al, 1996). Exquisite clinical judgment and good communication between members of the multidisciplinary team are crucial, because about 60% of hepatoblastomas are resectable at diagnosis.
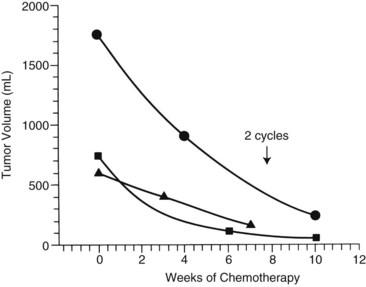
Complete resection of the primary tumor is necessary for survival and may require extended hepatic lobectomies or complex biliary reconstructions. For hepatoblastoma, reports have suggested that gross total resection of the primary lesion may be adequate for cure in chemoresponsive tumors (Dicken et al, 2004; Schnater et al, 2002).
For resected tumors (stage I) with fetal histology, further therapy is not recommended. All other stage I tumors without pure fetal histology, in addition to stage II patients, should receive four cycles of cisplatin, 5-fluorouracil (5-FU), and vincristine (C5V). Patients with stage III and IV disease should receive four cycles of chemotherapy, followed by resection or transplantation, followed by two more cycles of chemotherapy. C5V is administered, followed by doxorubicin if there is minimal response to C5V. Recent reports have suggested the use of doxorubicin from the start in this subgroup (Finegold et al, 2008; Malogolowkin et al, 2008). The combination of cisplatin plus doxorubicin was compared with cisplatin plus 5-FU plus C5V in a combined CCG/POG (intergroup) study (Ortega et al, 2000). The efficacy was thought to be similar, but more complications resulted with the regimen containing doxorubicin, accounting for equivalent event-free survivals; however, a more detailed review of the analysis suggested that the doxorubicin-containing arm had an improved disease-specific survival. This finding implied that with better management of toxicity, patient outcome might be better with a doxorubicin-containing regimen. In addition, a clinical trial that used a combination of cisplatin and carboplatin had to be terminated because of poor tumor response, suggesting that platinum-based monotherapy, as proposed by SIOPEL, was not as effective as combination chemotherapy. Clinical trials by the COG and SIOPEL are planned to evaluate the use of doxorubicin, irinotecan, and other agents, especially in high-risk patients.
In patients with unresectable primary tumors, the use of liver transplantation is increasing. A recent analysis stated an approximate 80% long-term disease-free survival in those receiving transplantation in large, solitary, or multifocal tumors invading all four sectors of the liver (Otte et al, 2005). The United Network for Organ Sharing (UNOS) database consists of over 200 patients, with a median age of 2.9 years, who underwent orthotopic liver transplantation (OLT) for hepatoblastoma between 1987 and 2006. Approximately half of the patients had a recurrence. Overall survival was 80%, 69%, and 66% at 1, 5, and 10 years, respectively (Austin et al, 2006). In a recent multicenter review, 147 patients with hepatoblastoma were analyzed after liver transplantation. In almost three quarters of these patients, the original surgery was OLT; the remaining patients either had residual disease after prior resection or had recurrent tumor. The first group of patients had an improved outcome, with 82% overall disease-free survival, compared with 30% in the second group. Smaller, single-center reports have reinforced findings that liver transplantation for hepatoblastoma has the best outcome when done as the primary procedure rather than as a salvage (D’Alessandro et al, 2007; Otte et al, 2004; Pham et al, 2007; Reyes et al, 2000).
Transplantation, however, does require the use of immunosuppressive treatment, which comes along with its own set of side effects. Moreover, there is in increased chance of thrombosis of the hepatic artery after transplantation in children (Jain et al, 2006). The main causes of mortality after transplantation, accounting for 54% of this population, are metastases and recurrence (Austin et al, 2006). The COG is continuing to investigate the role of liver transplantation in hepatoblastoma. A global database has been instituted to aid in this endeavor.
In one study, the 1-year survival for patients presenting with metastases was no different from that in patients with localized tumors (Van Tornout et al, 1997). In another study by SIOPEL, the 5-year overall and event-free survival for children with hepatoblastoma who presented with pulmonary metastases were 57% and 28%, respectively (Perilongo et al, 2000). This study suggested that 25% to 30% of patients with synchronous pulmonary metastases are curable. It is still necessary to resect the primary liver tumor, and pulmonary metastasectomy should be done only if the primary site is controlled (Schnater et al, 2002). Many pulmonary metastases resolve with chemotherapy, but thoracotomy and resection are sometimes required for larger or persistent metastatic lesions (Passmore et al, 1995).
No current prospective studies are underway for pulmonary metastasectomy, but one recent study describes the advantage of pulmonary metastasectomy for diagnosed lesions that remain after neoadjuvant therapy (Meyers, 2007). Some radiation oncologists have treated pulmonary metastases with external-beam radiotherapy in an approach similar to that used for Wilms tumors, but with 18 to 20 Gy administered (Habrand et al, 1992); however, this may be associated with significant pulmonary toxicity and has not resulted in cure. There is no reported cure in patients with spread to sites outside of the lung or local lymph nodes.
Outcome
Following a gross total resection, the 5-year event-free survival is 83%, but this drops to 41% in patients with tumor remaining after surgery (Ortega et al, 2000). Some patients with microscopic residual tumor are curable with continued chemotherapy and may benefit from external-beam radiotherapy to the primary hepatic site. Resection of many hepatoblastomas may be easier after chemotherapy, and complete resection of the primary hepatic tumor is necessary for survival. In multivariate analysis, factors that have been independent predictors of worse prognosis include a high TNM stage, unresectable tumor, bilobar involvement and multifocality, AFP less than 100 ng/mL or greater than 105 ng/mL, distant metastases, embryonal versus fetal histology, and vascular invasion (von Schweinitz et al, 1997b). The COG has reported a 3-year event-free survival of 90%, 50%, and 20% for stage I to II, III, and IV, respectively (Malogolowkin et al, 2008).
Future Directions
The uses of various novel treatments are currently being investigated. First, transcatheter selective arterial chemoembolization, which involves the direct injection of chemotherapeutic agents to the tumor, hypothetically decreases systemic toxicity. The average decrease in tumor size was 84% in one study (Clouse et al, 1994), and unresectable hepatoblastomas may become resectable with this intervention (Berthold et al, 1986). Doxorubicin, cisplatin, and fluorodeoxyuridine have been promising, as these agents have a high hepatic extraction; case reports exist with striking results. For example, Yokomori and colleagues (1991) describe the total regression of a tumor in a 4-month-old infant with fetal hepatoblastoma treated for 1.5 years with 5-FU, vincristine, doxorubicin, and cisplatin. No recurrence was seen after 6 years of follow-up. Risks involved with this technique include infection, thrombosis, or shifting of the catheter. Furthermore, it is challenging to carry out in children, and prospective studies are needed. Other new approaches include treatment with anti-AFP antibodies, IL-2, and viral transfection vectors to attack malignant hepatic cells (Geiger, 1996; Huber & Richards, 1996; Ji & St, 1997; Ramani et al, 1997).
Hepatocellular Carcinoma (Hepatoma)
Epidemiology
HCC (see Chapter 80) accounts for 23% of pediatric liver tumors but is rare in infancy (Finegold, 1994). Approximately 1.4 cases per 1 million children exist in the United States (Ni et al, 1997). The LCSG (1987) reported no cases in children age 4 years or younger in a series of 2286 patients with histologically reviewed tumors. Historic series without pathologic review may report a higher rate of infantile HCC owing to misdiagnosis of some early hepatoblastomas (Exelby et al, 1975). We have had personal experience of infants with well-documented HCC, and it affects about 0.5 children younger than age 15 years per 1 million annually. SEER data indicate that HCC accounts for 87% of primary hepatic malignancies in the 15- to 19-year age group (Darbari et al, 2003). The incidence is bimodal, with an early peak that is lower than that of hepatoblastoma. Most of these early cases occur before 5 years of age. A second peak occurs between 13 and 15 years of age. The median age at presentation of fibrolamellar carcinoma, a variant of HCC, is 20 years, and it is rarely observed before age 10 years. HCC has a male predominance (1.3 to 3.2 : 1), and in areas endemic for hepatitis B, the male/female ratio may be reversed, at 0.2 : 1. Approximately 35 to 40 new HCCs are diagnosed per year in the pediatric age group in the United States. Incidence reported for the years 1973 through 1977 versus 1993 through 1997 showed a decrease from 0.45 to 0.29 cases per 1 million (Darbari et al, 2003).
Hepatitis B and C infection correlates with the incidence of HCC. In Asia, 85% of HCC patients, both adult and pediatric, are hepatitis B surface antigen positive, whereas this is found in only 10% to 25% of patients in the United States. The relative risk for the development of HCC is 250 : 1 for patients with chronic active hepatitis compared with patients without hepatitis surface antigen positivity (Brower et al, 1998). Hepatitis C antibodies are found in 20% of patients with HCC. In one report, an infant with a history of neonatal hepatitis developed HCC (Moore et al, 1997a). Other conditions associated with the development of HCC include cirrhosis, α1-antitrypsin deficiency, tyrosinemia, aflatoxin ingestion, hemochromatosis, hepatic venous obstruction, androgen and estrogen exposure, Alagille syndrome (arteriohepatic dysplasia), and Thorotrast administration (Wegmann et al, 1996). One case of childhood HCC developing in a patient with neurofibromatosis has also been reported (Ettinger & Freeman, 1979).
A universal vaccination program against hepatitis B has reduced the incidence of HCC in Taiwan. The average annual incidence of HCC in children 6 to 14 years old declined from 0.70 per 100,000 children in the years 1981 through 1986 to 0.36 in the years 1990 through 1994 (P < .01) coincident with widespread administration of the hepatitis B vaccine (Chang et al, 1997). The mortality rate also decreased during this period. Antiviral therapy with lamivudine reduced the risk of development of HCC in patients with chronic hepatitis B infection and cirrhosis or fibrosis (Liaw et al, 2004). In contradistinction to HCC in adults, however, conditions associated with cirrhosis occur in only 30% to 40% of children with HCC; the remaining 60% to 70% of tumors are present without any cirrhosis (De Potter et al, 1987; Fattovich et al, 2004; Ismail et al, 2009; Llovet & Beaugrand, 2003; Marsh et al, 2004; Reynolds, 2001).
In one comparative study of pediatric HCC and hepatoblastoma, numerous discriminating features were reported (Chan et al, 2002). The mean age at presentation was 18 months for hepatoblastoma versus 10 years for HCC. The initial resectability of HCC was 45% and did not improve with chemotherapy, whereas 91% of hepatoblastomas could be completely resected before or after chemotherapy. Tumor rupture occurred in 36% of hepatoblastomas versus 9% of HCCs. Most importantly, the survival of patients with HCC was much worse.
Pathology (See Chapter 78)
HCCs are highly invasive and often multicentric at diagnosis with frequent hemorrhage and necrosis. Nuclear pleomorphism, nucleolar prominence, and the absence of extramedullary hematopoiesis are observed, and the cells are larger than normal hepatocytes. Low-grade HCCs may look similar to normal hepatocytes, especially if a limited amount of tissue is sampled. Invasiveness, and vascular invasion in particular, is a hallmark of these tumors. Extrahepatic dissemination to portal lymph nodes, lungs, and bones is frequent at diagnosis and strongly affects survival. HCCs naturally progress from capsular invasion to extracapsular extension, then vascular invasion, and finally to intrahepatic metastases (Toyosaka et al, 1996). A strong correlation has been found between intrahepatic metastases and portal vein thrombosis; this suggests that efferent tumor vessels anastomose to the portal, rather than the hepatic, veins, allowing intrahepatic spread and explaining the multicentricity that is a hallmark of HCC. Fibrolamellar HCC is a histologic variant seen in older children and young adults. It was once thought that this variant was associated with a better prognosis (Greenberg & Filler, 1989); however, studies indicate that when stage is controlled for, the survival is similar between standard HCC and its fibrolamellar variant.
Biology and Molecular Biology
Most investigations into the basic biology of HCC involve the study of hepatitis B and its relationship to carcinogenesis (Scaglioni et al, 1996). In one in vivo model in which rats developed HCC after prolonged feeding with glyceryl trinitrate, KRAS point mutations were identified in 8 of 18 animals that developed tumors (Tamano et al, 1996), and no TP53 mutations were seen. Cytogenetic data indicate that chromosomal abnormalities are complex, but no consistent patterns have been established (Terris et al, 1997).
Clinical Findings
Children and adolescents with HCC are often seen initially with palpable abdominal masses (40%), but many are symptomatic at diagnosis (Ni et al, 1997). Pain is frequent (38%) and may occur in the absence of an obvious mass. Constitutional disturbances such as anorexia, malaise, nausea and vomiting, and significant weight loss occur with greater frequency. Jaundice is an uncommon feature of either disease, but AFP is elevated in approximately 85% of patients, with most levels greater than 1000 ng/mL (Brower et al, 1998). Although elevated, these levels are usually less than those measured in hepatoblastoma patients; 10% may come to medical attention with tumor rupture and a hemoperitoneum (Brower et al, 1998).
Treatment
In over a quarter century, no significant progress has been made in treatment of the pediatric population with HCC (Ismail et al, 2009). This tumor remains extremely resistant to current chemotherapy agents, and long-term survival is impossible without complete resection. Because of a high incidence of multifocality within the liver, extrahepatic extension to regional lymph nodes, vascular invasion, and distant metastases, complete resection is often impossible. Infiltration with thrombosis of portal and hepatic venous branches is common, and even the vena cava may be involved. Furthermore, the cirrhosis found in a number of HCC livers may preclude an extensive resection. Most analyses report a 10% to 20% resectability rate among children with HCC. In contrast, patients with a fibrolamellar histologic pattern have a higher resection rate and essentially do not have cirrhotic livers. Even with complete resection, however, the prognosis remains poor secondary to the high rate of recurrence.
Historically, the same chemotherapy protocols used for hepatoblastoma were also applied to HCCs in childhood; however, HCC is rather unresponsive to chemotherapy overall, although cisplatin in particular has shown activity against it (Bower et al, 1996
Related posts:
 Preoperative and postoperative nutrition in hepatobiliary surgery
Medical management of bleeding varices: Primary and secondary prophylaxis
Preoperative and postoperative nutrition in hepatobiliary surgery
Medical management of bleeding varices: Primary and secondary prophylaxis
 Portal hypertension in children
Portal hypertension in children
 Chemotherapy and radiotherapy for pancreatic and periampullary cancer: Adjuvant, neoadjuvant, and palliative
Chemotherapy and radiotherapy for pancreatic and periampullary cancer: Adjuvant, neoadjuvant, and palliative
 Bile duct exploration and biliary-enteric anastomosis
Bile duct exploration and biliary-enteric anastomosis
 Interventional endoscopy: Technical aspects
Interventional endoscopy: Technical aspects
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


