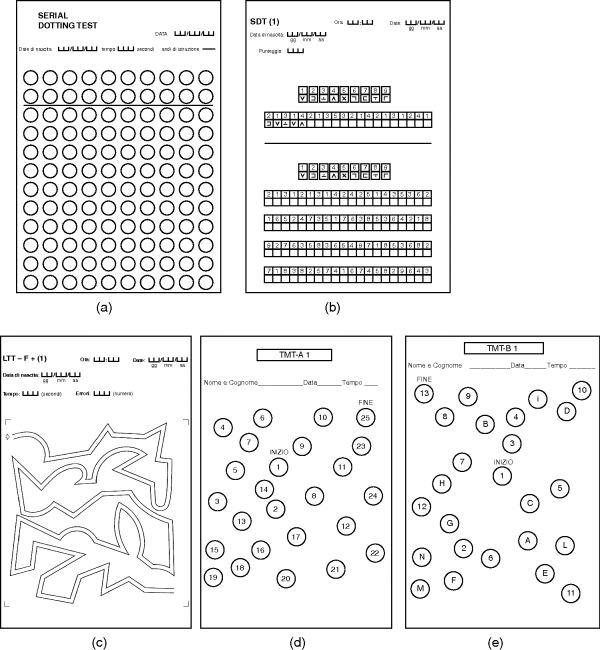
Chapter 11 Piero Amodio Department of Medicine (DIMED) University of Padova, Padova Italy Cirrhosis, far from being an isolated disorder of the liver, has well known consequences on the whole body and, notably, on brain functioning. The influence of liver failure on the brain is clearly evidenced by mental and/or behavioral changes that have been known since Ancient Greek and Roman times. Hippocrates recognized a “symptomatic delirium” related to “jaundice and suppression of natural periodical evacuation” [1]. In the last centuries, the issue of mental and behavioral alterations in individuals with advanced liver disease was studied by many authors [2,3]. The neurological or psychiatric alterations occurring in severe liver failure were termed portosystemic encephalopathy (PSE) [4] and later included in the term hepatic encephalopathy (HE) [5]. HE can be defined as brain dysfunction caused by liver insufficiency and/or portosystemic shunting that produces a spectrum of neurologic and psychiatric abnormalities ranging from subclinical alterations to coma [6]. Consequently, the encephalopathies caused by isolated defects of liver metabolism (e.g., urea cycle disorders, Reye’s syndrome) or valproate hyperammonemia are not implicated in the term HE. HE produces a wide and nonspecific spectrum of neurological and psychiatric abnormalities. In its mildest form, HE is detectable only by psychometric or neurophysiologic tools. It manifests clinically with personality changes, such as apathy or irritability, and inappropriate behavior that may be reported by patient’s relatives; also, obvious mental changes and motor function are detectable on clinical examination. Mental changes progressively include disorientation to time and space, inappropriate behavior, delirium with agitation or somnolence, stupor and, finally, coma. Of motor changes, asterixis or “flapping tremor” is the most typical and can be detected in the early middle stages of HE, but not in coma. Actually, asterixis is not tremor, but negative myoclonus consisting of loss of postural tone that is easily elicited by the hyperextension of the wrists with separated fingers. However, it may involve other muscles. Rarely, transient focal neurologic signs may occur in severe HE [7]. In some patients, massive motor alterations occur. The label of non-Wilsonian hepatocerebral or hepatolenticular degeneration was given to persistent HE with prominent extrapyramidal and/or pyramidal signs in which postmortem brain examination proves the existence of relevant pathologic changes with brain atrophy [8]. Rare cases of paraplegia with progressive spasticity and weakness of lower limbs with hyperreflexia and relatively mild persistent or recurrent mental alterations that may or may not reverse with liver transplantation [9,10] are preferentially labelled as hepatic myelopathy [11]. Sleep disturbances were reported in subjects with cirrhosis [4,12,13]; daily somnolence is the one that has a proven relationship with HE [14]. Finally, it was recently recognized that HE is associated with falls; if this is caused by cognitive or motor abnormalities or by their combination, or even by cirrhosis per se, is not yet clear [15,16]. In brief, the manifestation of HE can be summarized into the five patterns in Table 11.1. The exact grading of the severity of mental alterations of HE is less relevant than the pattern of HE presentation, because this latter is of great importance both for HE recognition and for differential diagnosis. Staging of the severity of mental alteration is simple and can be easily done according to West Haven criteria (Table 11.2), possibly adopting operative criteria [17]. Table 11.1 Clinical patterns of hepatic encephalopathy presentation. Table 11.2 Grading of the severity mental alterations in hepatic encephalopathy. Recently, the term “covert” HE was coined for patients with grade 1 HE (barely discernible on clinical examination) or minimal HE (i.e., only detectable by specific tools) (Table 11.2) [18]. Other grading systems can be used (CHESS scale [19], HESS scale [20], Glasgow Coma Scale, Mo-log [21]), but there is no clear proof that they are better than the widely used West Haven classification, once operative definitions of staging are used. Proper assessment of HE takes into account not only grading of the severity of mental alteration, but also the underlying condition producing HE, the time course of HE, and the existence of precipitating factors. Therefore, proper assessment of HE is multi-axial [24] (Table 11.3). Therefore, to properly HE in a single patient, all the four items should be mentioned. If possible, the information on treatment should be added, because this can provide cues for diagnosis (a mental disorder absolutely unresponsive to treatment for HE may be something other than HE) and for alternative or add-on treatment. For instance, a proper report for a single patient with a bout of HE is “type C episodic overt HE (grade 3) precipitated by paracentesis in a subject on lactulose treatment.” Table 11.3 The multi-axial classification of hepatic encephalopathy (HE). The prevalence of HE is related to the severity of liver dysfunction and/or the extent of portosystemic shunting. The prevalence of overt HE is 10–14% in patients with cirrhosis and 16–21% in those with decompensated cirrhosis [25,26]. The prevalence in noncirrhotic portal hypertension is not defined. The prevalence of minimal HE varies according to: (i) the occurrence of previous bouts of overt HE [27,28]; and (ii) the tools used for its detection, because it escapes obvious identification. A conservative esteem is about 30% in inpatients with cirrhosis. The incidence of overt HE was 9% at 5 years and 25% at 10 years in a cohort of subjects with virus-related hepatitis after the first diagnosis of cirrhosis [29]. Another study reported 25% cumulative incidence of HE at 5 years after the diagnosis of compensated cirrhosis of various etiologies [30]. Patients with decompensated viral cirrhosis have 38% risk of overt HE within 2 years [31]. Subjects with a previous bout of overt HE have a 40% cumulative risk of another bout of overt HE at 1 year [32], and subjects with more than one bout of overt HE in the previous 6 months have a 40% cumulative risk of another bout within 6 months, despite disaccharide treatment [33]. In individuals with minimal HE detected by cognitive or EEG criteria, the incidence of bouts of overt HE is 26–35 per 100 patient-years [34,35]. Subjects at higher risk for HE are those who underwent transjugular intrahepatic portosystemic shunt (TIPS) or surgical portosytemic shunt. The 1-year risk for HE after the procedure is estimated to be about 10–50% [36,37]. At any rate, the incidence of HE after TIPS and after surgical portosystemic shunt [38] is greatly influenced by selection criteria [39]. HE accounts for approximately 100,000 hospitalizations each year in the United States [40]. The diagnosis of HE is not always as simple as might be expected and this depends, in part, on poor insight into the concept of HE. This can lead to errors, ascribing uncritically to HE any neuropsychiatric disorder occurring in patients with liver disease, even without the evidence of a causal link to liver failure or portosystemic shunting. Consequently, brain disorders that only occasionally coexist with liver disease can be mislabeled as HE, preventing patients from receiving proper etiologic treatment. Basically, the diagnosis of HE is based on the clinical findings; the differential diagnosis is based on the Bayes’ theorem, which states that the probability of a disease is given by: (i) the a priori probability that the disease may exist; and (ii) the likelihood of symptoms and/or signs. The probabilities of the occurrence of different diseases should then be compared. Therefore, the first step towards a diagnosis of HE is the detection of neurologic or psychiatric findings compatible with HE. The second step is the recognition that the patient has severe liver insufficiency and/or portosystemic shunting, because these conditions provide the a priori probability of HE. The risk of HE is related to the severity of liver dysfunction, with the exception of the subgroup of individuals with large portosystemic shunting, as these risk factors are independent [41,42], so that patients with huge shunting can experience HE even without liver insufficiency. Hyperammonemia is a good marker of portosystemic shunting, even with negligible liver disease [43], because ammonia has high first-pass liver metabolism (about 70%) [44,45] and its plasma level increases if portal liver perfusion is reduced due to portosystemic shunting [46,47]. The value of hyperammonemia as a prerequisite for the occurrence of HE was emphasized by Conn [48], but was later neglected or even disputed [49], possibly due to confusion between two concepts: In fact, even admitting that the pathophysiology of HE is mutifactorial or uncertain, it is hardly conceivable that a patient with low plasma ammonia may have encephalopathy caused by liver disease and/or portosystemic shunting. In patients with low plasma ammonia, due to the very low a priori probability of HE, intense scrutiny for alternative causes of mental alteration is mandatory. In addition, recently it was clearly proven that high ammonia plasma level is a risk factor for the development of HE [50]. Of note, ammonia determination needs some precaution, because ammonia develops rapidly in blood maintained at room temperature. Therefore, blood samples should be immediately put in ice, rapidly delivered to the laboratory, and the determination should be performed as soon as possible. The use of venous blood is simpler, but arterial or capillary blood may be preferable. The third step is the recognition of precipitating factors for HE (e.g., bleeding, constipation, and infections) or a history of previous bouts of HE, which further increase the risk of HE. However, HE can occur without their occurrence. The fourth step is the exclusion of alternative causes or the detection of concomitant causes of brain alteration, depending on the clinical findings. However, the exclusion of alternative causes does not require always formal investigation because it can be obtained by the history and clinical manifestations together with probabilistic reasoning. Finally, the response to treatment should be considered to confirm the diagnosis or suggest the existence of alternative or concomitant neurologic disorders (Figure 11.1). Figure 11.1 Flow chart for the diagnosis of hepatic encephalopathy (HE). ARD, ammonia reducing drugs (disaccharides/rifaximine or other antibiotics). On closer inspection, for the patient with coma or confusion, the differential diagnosis between HE and other disorders is reported in Table 11.4 For the patient with almost continuous mild mental alterations interspersed with relapses of confusion or even coma, the differential diagnosis between relapsing and persistent HE and other disorders is reported in Table 11.5. Finally, for the patient with prominent motor disorder and mental/behavioral alterations, the differential diagnosis between HE and other disorders [51] is reported in Table 11.6. Table 11.4 Differential diagnosis of HE for patients with acute confusion or coma. Table 11.5 Differential diagnosis of HE for patients with highly relapsing/persistent mental impairment. Table 11.6 Differential diagnosis of HE for patients with prominent motor disorder with mild/moderate mental decay. Considering these alterative disorders that should be taken into account in the differential diagnosis of HE, the diagnostic procedures that are required for the diagnosis of patients with suspected HE are reported in Table 11.7. Particular problems are: (i) the association; and (ii) the interaction between HE and other neurologic and metabolic disorders that can produce encephalopathy per se, but can also interact in the pathophysiologic pathways producing HE. An example of the former is HE occurring in an individual who is already suffering from vascular dementia (stroke sequelae, Binswanger’s disease), Alzheimer’s disease, or post-traumatic brain sequelae. The clinical improvement – albeit incomplete – after treatment for HE orients towards a diagnosis of HE associated with another brain disease. An example of the latter is sepsis that can produce encephalopathy per se (septic encephalopathy) [52], but also interacts with the mechanisms producing HE and is therefore a cause of HE [53]. The same occurs for hyponatremia [54,55] and, possibly, thiamine deficiency [56]. Table 11.7 Diagnostic procedure for patients with suspected HE. The existence of concurrent causes of brain dysfunction should be considered, diagnosed, and properly treated, because mixed encephalopathies do exist and neglecting to treat one of them can have deleterious consequences (e.g., coexisting thiamine deficiency). The response to treatment to disaccharides and rifaximin provide information on the role that HE can have in patients with overlapping disorders (e.g., Alzheimer disease or vascular dementia and HE) (Figure 11.1). HE can occur without clinical evidence (minimal HE, MHE) or, at least, without clear disorientation (covert HE, CHE). Brain alteration in these patients is proven by neuropsychologic or neurophysiologic investigation. Cognitive alterations mainly regard attention, working memory, visuomotor abilities, speed in fine movements, and executive function. In contrast, verbal abilities and verbal memory, both short-term and long-term memory are preserved or relatively preserved, at odds with the amnestic mild cognitive impairment that frequently heralds Alzheimer’s disease. As memory and verbal alterations are more easily noted by relatives and physicians, than attention disturbance subjects with MHE can be easily missed on routine clinical examination. Paper and pencil tests that are suited for the detection of MHE are the ones comprising the Portal-Hepatic Psychometric Score (PHES; i.e., the Trial Making Test A, TMT-A) – also called number connection test (NCT) – the Trial Making Test B (TMT-B), the Digit Symbol test (DST), the Serial Dotting Test (SDT), and the Line Trait test (LTT) (Figure 11.2) [57,58]. Other tests exploring the same areas, such as the Block Design Test or the Figure Connection Test can be used; the latter can substitute for TMT-B in individuals who do not know the alphabet [59–61]. The use of more than one test is recommended [24]. There is consensus that the PHES is the preferable diagnostic tool based on paper and pencil tests [62]. It can be simplified using only three tests (i.e., DST, SDT, and LTT) [63]. Computerized tests exploring working memory or inhibition and attention [17,64,65] have also proven to be useful. They require some familiarity with computer usage. Figure 11.2 Pencil and paper psychometric tests useful for the detection of minimal hepatic encephalopathy (MHE). A, Serial Dotting Test; B, Digit Symbol Test; C, Line Trait Test; D, TMT-A; and E, TMT-B (Italian forms, based on Italian alphabet). The use of any neuropsychologic tests (paper and pencil or computerized) for the diagnosis of MHE require that: In addition, it should be noted that psychometric information, as well as other functional data, is nonspecific. Therefore, other clinical conditions causing mild cognitive impairment should be considered in differential diagnosis (Table 11.8). Table 11.8 Differential diagnosis for minimal and covert HE.
Hepatic Encephalopathy
Introduction
Definition
Clinical Manifestation
Pattern
Description
Coma
The patient is eye closed, unresponsive even to pain stimulation
Abrupt confusional state
Inhibited
The patient is disoriented in time or/and space or/and identity and somnolent
Agitated
The patient is disoriented in time or/and space or/and identity and agitated/angry/restless
Almost continuous mental dysfunction with interspersed recurrent confusional episodes
The pattern is dementia-like
Predominant motor disorder with mild/moderate mental dysfunction/confusion
Extrapyramidal
Parkinsonism, chorea, or athetosis
Pyramidal
Spastic paraparesis with hyperyflexia
Minimal brain dysfunction
The patient is oriented and his/her mental activity seems perfectly normal or nearly normal; however, on psychometric testing cognitive alterations are detectable (concerning attention, working memory, visuopractical ability, inhibition). Other signs are the slowing of EEG activity and/or the reduction of critical flicker frequency
ISHEN criteria [18]
West Haven grading [22], updated concerning the conditions “MHE” and “unaffected”
Operative criteriab
Unaffected
Covert
Minimal
Psychometric and/or EEG alterations without mental changes on routine clinical examination
Grade 1
Trivial lack of awareness
Euphoria or anxiety
Shortened attention span
Impairment of addition or subtraction
Lack of consensus on definition, low inter-rater reproducibility
Overta
Grade 2
Lethargy or apathy
Disorientation for time
Obvious personality change
Inappropriate behavior
Disorientation for time
(≥ three incorrect items)
With or without other symptoms
Grade 3
Somnolence to semi-stupor
Responsive to stimuli
Confused
Gross disorientation
Bizarre behavior
Disorientation for placec
(≥ three incorrect items)
Plus Disorientation for time
With or without other symptoms
Grade 4
Coma, unable to test mental state
aThe presence of asterixis (flapping tremor) is considered an independent marker of overt HE; note that it is absent in coma.
bOperative criteria were suggested to reduce misclassification caused by the colloquial and poorly defined terms used in West Haven classification [17].
cAt variant from Folstein et al. [23], three items are required for disorientation in space, because hospitalized patients may reasonably ignore the floor.
Domain/dimension
Name of the domain
Subdivision
Description
Underlying condition producing HE
Type
A
Caused by ALF
B
Caused by PS without significant liver disease
C
Caused by cirrhosis (both liver disease and PS shunting)
Severity of mental alterations
Grade
MHE
Covert HE (see Table 11.2)
1
2
Overt HE (see Table 11.2)
3
4
Interval between bouts of HE
Time course
Episodic
An occasional bout of HE
Recurrent
Bout of HE occurring ≤6 months from the previous one(s)
Persistent
Patient is always more or less symptomatic
Presence of precipitating factors
Precipitation factors
Precipitated
There is an obvious factor that produced the occurrence of the bout of HE. If this the case it should be mentioned
Not precipitated
Absence of a known factor to which to ascribe the occurrence of the bout of HE
ALF, acute liver failure; MHE minimal hepatic encephalopathy; PS, portosystemic shunting.
Epidemiology
Diagnostic Tools and Differential Diagnosis
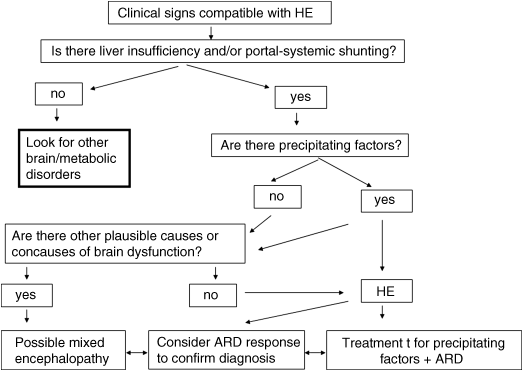
For agitated confusion state, consider drug intoxication and acute psychosis
*Sepsis, hyponatremia, and, to a lesser extent, thiamine deficiency are also implicated into the pathophysiology of HE, so that the conditions may overlap.
Clinical history
Clinical examination for hepatic stigmata, asterixis, and exclusion of neurologic focal signs, rigor, evidence of trauma
Body temperature (fever, hypothermia), blood pressure, pulse frequency, chest and abdomen clinical examination
Blood sample for:
Urine examination for WBC (in confusion/coma)
Polymorphonuclear leukocyte count in ascites, if there is ascites (in confusion/coma)
Brain CT scan (in sudden coma, focal signs, or rigor)
EEG (if nonconvulsive epilepsy, herpes encephalitis, or malingering are suspected)
Brain MRI (in dementia-like syndromes) + spinal MRI with FLAIR sequences (in prominent motor manifestations)
Cerebrospinal fluid examination (if meningitis or encephalitis are suspected)
Motor EP, electromyography, somatosensory EP (in prominent motor manifestations)
Splanchnic US Doppler study and/or CT scan with portal system reconstruction (in highly recurrent HE, persistent HE, and in HE with relevant motor component)
EP, evoked potential; INR, international normalized ratio; WBC, white blood cell.
Diagnosis of Minimal and Covert HE
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


