Fig. 13.1
Oxidative pathways of ethanol metabolism . ADH alcohol dehydrogenase, ALDH aldehyde dehydrogenase, NAD nicotinamide adenine dinucleotide, NADH reduced NAD, NADP nicotinamide adenine dinucleotide phosphate, H 2 O 2 hydrogen peroxide, ROS reactive oxygen species
Consequences of Alcohol Metabolism by Oxidative Pathways: Cancer Implication
Alcohol metabolism in the liver results in various products/effects with implications for hepatocarcinogenesis.
Acetaldehyde Generation/Adduct Formation
Acetaldehyde produced by alcohol oxidation, if accumulated to appreciable concentrations, can form adducts with DNA and RNA and decrease DNA repair. Acetaldehyde also has the capacity to react with lysine residues on proteins including enzymes, microsomal proteins, and microtubules and affect their function. Formation of protein adducts in hepatocytes may contribute to impaired protein secretion, resulting in hepatomegaly. In addition, there is evidence that acetaldehyde and malondialdehyde (a by-product of lipid peroxidation) can combine and react with lysine residues on proteins, giving rise to stable malondialdehyde–acetaldehyde (MAA) protein adducts that can be immunogenic and, thus, can contribute to immune-mediated liver damage. Also, MAA adducts have proinflammatory and profibrogenic properties.
The most relevant acetaldehyde adducts that impact the genome function and have implications to carcinogenesis are their interaction with the exocyclic amino group of deoxyguanosine to form DNA adducts (Fig. 13.2). These adducts involve the reaction of one molecule of acetaldehyde with DNA to form N2-ethylidenedeoxyguanosine, which is relatively unstable and abundant in human liver even in the absence of exogenous acetaldehyde [11]. This adduct is reduced in vivo with glutathione or vitamin C to form the stable N2-ethyldeoxyguanosine (Et-dG). In addition, two molecules of acetaldehyde, or crotonaldehyde, form an adduct known as N2-propano-2′-deoxyguanosine [12], which is maintained at a low steady state by DNA repair. A secondary acetaldehyde-related DNA adduct, 1,N2-etheno-dG, is formed from acetaldehyde-stimulated lipid peroxidation. For a detailed discussion of these adducts and their genotoxic effects, the reader is referred to the review article by Brooks and Zakhari [13].
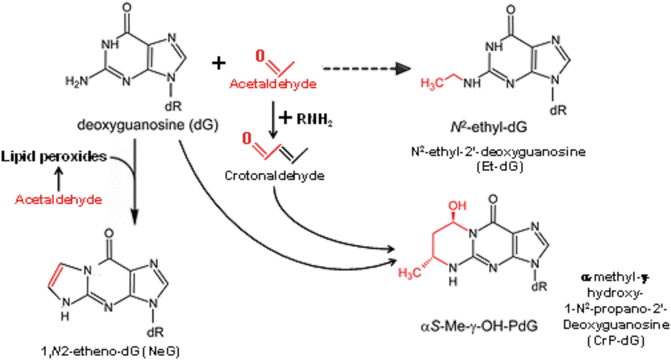
Fig. 13.2
Acetaldehyde : DNA adduct formation
It should be noted that some results obtained from ADH1B polymorphisms do not concord with the acetaldehyde hypothesis. For example, a decreased UADT cancer risk was observed in drinkers who carried the ADH1B*2 allele that codes for the more active enzyme, leading to high acetaldehyde exposure [14].
Formation of Reactive Oxygen Species, Reactive Nitrogen Species, and Oxidative Stress
Hepatic mitochondria produce ROS through the activity of the electron transport chain (ETC) as a by-product of oxidative phosphorylation. Normally, a small fraction of electrons entering the ETC can prematurely escape from complexes I and III and directly react with ~1–3 % of respiratory oxygen molecules to generate the superoxide anion radical, which is then dismutated by the mitochondrial manganese superoxide dismutase (MnSOD) into hydrogen peroxide (H2O2). Mitochondrial glutathione peroxidase (GPx) then converts H2O2 into water by using reduced glutathione (GSH) as a cofactor. Thus, most of the ROS generated by the ETC in the normal state are detoxified by the mitochondrial antioxidant defenses. The non-detoxified portion of ROS diffuses out of mitochondria and affects signal transduction pathways and gene expression, triggering cytokines, hormones, and growth factors, which if excessive may lead to hepatic inflammation, necrosis, and/or apoptosis. In addition, metals (e.g., iron and copper) can further react with H2O2 to produce hydroxyl radicals via the Fenton reaction. Nitric oxide (NO) , a reactive nitrogen species critical for hepatocyte biology, can interact with peroxides to generate peroxynitrite (ONOO−), which, depending on the amount and duration, could be detrimental to the liver (Table 13.1). NO is produced from l-arginine and oxygen by iNOS, which is expressed in all liver cells (hepatocytes, stellate cells, Kupffer cells, and vascular endothelial cells), and its expression is induced by IL-1β alone or in combination with TNF-α, IFNγ, and/or LPS. Alcohol not only produces ROS and reactive nitrogen species (RNS), but also depletes antioxidants in cells resulting in “oxidative stress.” This condition regulates both genetic and epigenetic cascades underlying altered gene expression in human cancer [15].
Table 13.1
Chemical equations relevant to reactive oxygen and reactive nitrogen species generation
Reactive oxygen species generation O2 + e− → O2−• (superoxide anion) O2 − + H2O → HO2 • (hydroperoxyl radical) HO2 • + e− + H → H2O2 (hydrogen peroxide) H2O2 + e− → OH− + •OH (hydroxyl radical) |
Reactive nitrogen species generation l-arginine + O2 → •NO (nitric oxide) + l-citrulline O2•− + •NO → ONOO− (peroxynitrite) |
Fenton reaction (catalyzed by transition metals) H2O2 + Fe2+→ OH− + •OH + Fe3+ Haber–Weiss reaction (catalyzed by transition metals) H2O2 + O2 −• → O2 + •OH + OH− |
It has been suggested that ROS participate in tumor progression by promoting DNA damage and/or altering cellular signaling pathways [16]. A reliable biomarker of oxidative stress and ROS-induced carcinogenesis is 8-oxo-7,8-dihydroguanine (8-oxoGua), which is strongly implicated in all stages of carcinogenesis [17]. Formation of 8-oxoGua lesions has been shown to induce DNA base mutations in the TP53 tumor suppressor gene in liver cancer cells [18]. Elevated levels of 8-OHdG in transgenic mice infected with HBV can lead to development of hepatocellular carcinoma [19]. In addition, oxidative stress often renders repair mechanisms ineffective. ROS have been reported to promote hypermethylation of the promoter region of the tumor suppressor E-cadherin, a regulator of the epithelial-to-mesenchymal transition, in HCC cells [20]. ROS induce hypermethylation of the E-cadherin promoter by increasing Snail expression. Alcohol promotes breast and colon cancer progression through stimulating the EMT program via a Snail-mediated pathway [21] and may have a similar effect in HCC. Exacerbating oxidative stress in livers infected with HCV by ROS induction and by hampering the antioxidant system facilitates hepatocarcinogenesis [22]. Ironically, increases in Nrf2 protein, a transcription factor that regulates important antioxidant and phase II detoxification genes, were observed in hepatocytes of alcohol-fed mice, suggesting that Nrf2 plays a key role in the adaptive response against increased oxidative stress caused by CYP2E1 [23].
Increase in NADH/NAD+ Ratio
Alcohol metabolism produces a significant increase in the hepatic NADH/NAD+ ratio in both the cytosol and the mitochondria, as evidenced by an increase in the lactate/pyruvate and β-hydroxybutyrate/acetoacetate ratios, respectively [24] Consequently, alcohol oxidation vastly increases the availability of oxidizable NADH to the electron transport chain in the mitochondria. The liver responds to alcohol exposure in part by increasing the rate of oxygen uptake, which may lead to periods of hypoxia, particularly in the downstream (pericentral) parts of the liver lobule. Increased NADH/NAD+ ratio provides reducing equivalents and thus enhances the activity of the respiratory chain, including heightened oxygen use and ROS formation [25]. Furthermore, the increase in the NADH/NAD+ ratio results in derangement of carbohydrate metabolism and modulation of gene expression of, among others, SIRT1, a NAD+-dependent deacetylase [26] whose substrates include histones and the transcription factor p53 [27]. Increased NADH in hepatocytes due to alcohol metabolism may promote tumor growth by favoring the generation of lactate through a NADH-dependent enzyme, lactate dehydrogenase A (LDH-A), which catalyzes the conversion of pyruvate to lactate during glycolysis. Tumor cells utilize more glucose than normal tissue, favor aerobic glycolysis, and rely on lactate production for their survival. A molecular mechanism underlying the enhanced lactate production in cancer cells involves tyrosine phosphorylation which enhances LDH-A enzyme activity to promote tumor growth by regulating the NADH/NAD redox homeostasis [28].
Derangement of Metabolic Pathways
Increased NADH/NAD+ ratios in both the cytosol and mitochondria of hepatocytes influence the direction of several reversible reactions leading to alterations in hepatic lipid, carbohydrate, protein, lactate, and uric acid metabolism. These changes include (1) alcoholic hypoglycemia , the increase in NADH prevents pyruvate conversion to glucose by lowering the concentration of pyruvate, which in turn decreases the pyruvate carboxylase reaction, one of the rate-limiting steps of gluconeogenesis; (2) hampering of the tricarboxylic acid (TCA) cycle function , the increase in mitochondrial NADH in hepatocytes contributes to the saturation of NADH dehydrogenase; and (3) alcoholic acidosis, ketoacidosis is common in chronically malnourished alcoholics and is due to the formation of ketone bodies, primarily β-hydroxybutyrate [29]. In addition, the increase in NADH favors the conversion of pyruvate to lactate, resulting in lactic acidosis. The increase in NADH/NAD+ ratio diminishes pyruvate dehydrogenase (PDH) activity in the mitochondria, resulting in diminished conversion of pyruvate to acetyl-CoA. PDH activity is further diminished in chronic alcoholics due to hypomagnesemia and thiamine deficiency, resulting in the inhibition of pyruvate utilization in the TCA cycle; (4) hypoxia, alcohol metabolism by hepatocytes tends to increase oxygen uptake, resulting in significant hypoxia in the perivenous hepatocytes, the site of early liver damage due to chronic alcohol consumption.
Perhaps the derangement most relevant to HCC is alcohol impairment of retinoic acid (RA) synthesis and transport. Alcohol dramatically changes vitamin A and RA availability [30] and thus can impact carcinogenesis [31]. RA deficiency in alcoholics results from poor dietary intake and decreased absorption of retinoids and the significant overlap in metabolic pathways of alcohol and retinol, the alcohol form of vitamin A. Alcohol and retinol can be oxidized by similar, and sometimes identical, enzymes. In addition to the competitive inhibition of RA biosynthesis, prolonged alcohol consumption decreases tissue RA concentrations by enhancing its catabolism through the induction of cytochrome P450 enzymes and increasing mobilization of retinoids from the liver to extrahepatic tissues [32]. As a result, alcohol profoundly depletes hepatic retinoids and alters their distribution in other tissues [33, 34]. In addition to directly affecting RA metabolism and transport, alcohol may also affect plasma retinol concentration and its organ distribution indirectly through LPS-induced inflammation that reduces the level of RBP mRNA in the liver resulting in the impairment of the transport of retinol from the liver to plasma [35].
Changes in RA availability due to dysregulation of retinoid transport by alcohol are also becoming more evident [1]. It is strongly linked to alterations in differentiation/proliferation status of hepatocytes.
RA acts as a signaling molecule and regulates gene expression by binding to two subclasses of nuclear receptors, retinoic acid receptors (RARα, β, and γ isotypes) and retinoid X receptors (RXRα, β, and γ isotypes), encoded by distinct genes [36]. Alcohol affects the expression and activation of RA receptors, which in turn can impair the signaling events and induce harmful effects on cell survival and differentiation [37]. Recent developments indicate that alcohol can contribute to the aberrancy of retinoid nuclear receptor function and increased risk of cancer development through epigenetic alterations [38]. Alterations in the level of expression or functional activity of retinoid nuclear receptors are associated with a variety of cancers despite normal vitamin A levels. Hepatic retinoid level reduction by alcohol can lead to enhanced fibrogenesis that, in turn, may eventually constitute an irreversible process with regenerative diffuse parenchymal nodular transformation, cirrhosis, and HCC. Alcohol-related HCCs are associated with cirrhosis in a majority of cases, indicating that the pathological events leading to cirrhosis precede those causing cancer or that the structural alterations of cirrhosis favor hepatocyte dedifferentiation [39]. Since dedifferentiation is ultimately associated with increased proliferation rate, the dedifferentiation hypothesis fits perfectly with findings that low hepatic RA concentration due to alcohol leads to an upregulation of AP-1 (c-jun and c-fos) and beta-catenin-dependent gene expression that may promote proliferation and malignant transformation of hepatocytes by alcohol [34, 40]. Interestingly, alcohol-induced RA-dependent hepatocyte hyperproliferation may not only lead to the neoplastic transformation of preexisting hepatocytes but may also compromise organ regeneration by liver stem cells. The activation of liver stem cells requires a uniform inhibition of parenchymal proliferation [41] and their differentiation depends on RA [42, 43]. It is interesting to note that liver stem cells are highly responsive to vitamin A deprivation [44] and always appear in close proximity to activated hepatic stellate cells (HSC) suggesting a possible involvement of RA in control of their behavior. Deficiency of RA in alcoholic livers blocks differentiation and apoptosis in the progeny of liver stem cells, while promoting their proliferation. This may explain the development of anaplastic poorly differentiated HCCs without preexisting cirrhosis, which are also observed in alcoholics, albeit rarely [45, 46].
Variations in Metabolic Enzymes
Class I ADH and ALDH2 play a central role in alcohol metabolism. Allelic variations in the genes encoding ADH and ALDH produce alcohol- and acetaldehyde-metabolizing enzymes that vary in activity. These genotypes modify the susceptibility to tissue damage. The ADH gene family encodes for enzymes that metabolize various substrates, including retinol, and are differentially expressed in different organs. This highlights the important issue of substrate competition in alcohol-induced tissue damage. Genetic polymorphism occurs at the ADH1B and ADH1C loci [47] with different catalytic activities for alcohol. The ADH1B alleles occur at different frequencies in different populations. For example, the ADH1B*1 form is found predominantly in Caucasian and Black populations, while ADH1B*2 frequency is higher in Chinese and Japanese populations and in 25 % of people with Jewish ancestry. A significant interaction exists between ADH1B polymorphism and heavy alcohol consumption especially for those with ADH1B*1/*1 genotype and esophageal [48] and UADT [49] cancer. Several isozymes of ALDH have been identified, but only the cytosolic ALDH1 and the mitochondrial ALDH2 metabolize acetaldehyde. There is one significant genetic polymorphism of the ALDH2 gene, resulting in allelic variants ALDH2*1 and ALDH2*2 (glutamine to lysine substitution at position 487, resulting in 100-fold increase in the Km for NAD+, making the gene product virtually inactive). The low activity ALDH2*2 is a deficient phenotype, which is present in about 50 % of the Taiwanese, Han Chinese, and Japanese populations [50] and shows virtually no acetaldehyde-metabolizing activity in vitro. The activity of ADH and ALDH isozymes contributes to alcohol-induced tissue damage. Alcoholic cirrhosis is reduced over 70 % in populations carrying the ALDH2*2 allele [51]. The activities of class I ADH are much higher in cancerous than in healthy tissues [52], and individuals with the ADH1C*1 allele have an increased risk to develop breast cancer from alcohol [53]. Furthermore, ALDH2-deficient individuals are at much higher risk of esophageal cancer (specifically squamous cell carcinoma) from alcohol consumption than individuals with fully active ALDH2 [54]. The correlation between genetic polymorphism of ADH and ALDH and esophageal, head, and neck cancers was reviewed by Yokoyama and Omori [55] and summarized in Table 13.2.
Table 13.2
Alcohol, genetic polymorphism, and cancer
Population | Cancer type | Genotype | Result | Reference |
|---|---|---|---|---|
Asian | UADT | ADH1B*1 (slow) | Increased risk | |
Asian | UADT | ALDH2*1/*2 | Increased risk | |
Asian | Esophageal | ADH1B*1/*1 | Increased risk | [48] |
Japanese | Esophageal | ALDH2*1/*2 | Increased risk | [158] |
European | Various | ADH1B*2 (fast) | Protective | Hashibe et al. (2006) |
Caucasians | Head and neck | ADHIC*1 (fast) | Increased risk | |
European | Head and neck | ADHIC*2 (slow) | Increased risk | Hashibe et al. (2006) |
Caucasian | Head and neck | ADHIC*2 (slow) | Increased risk | |
Japanese | Head and neck | ALDH2*1/2 | Increased risk | [163] |
Although several CYP2E1 polymorphisms have been identified, only a few studies were undertaken to determine the effect on alcohol metabolism and tissue damage. In one study, the presence of the rare c2 allele was associated with higher alcohol metabolism in Japanese alcoholics but only at high blood alcohol concentrations of 0.25 g/dL [56]. In addition, induction of CYP2E1 also contributes to carcinogenesis through activation of pro-carcinogens, such as nitrosamines present in diets and in tobacco smoke, to their carcinogenic metabolites [57]. The correlations between genetic polymorphisms and risk of alcohol-related cancers are reviewed elsewhere [58].
Epigenetic Modifications
Functional genomic studies (GWAS, whole-genome sequencing, global DNA copy numbers and methylation, and gene or noncoding RNA expression profiling) revealed that genetic polymorphisms of immune-related genes, such as IL28B and MHC class I and II molecules (e.g., MICA), and somatic mutations of TP53 and ARID2 (a novel liver cancer-related gene that encodes a component of the SWI/SNF chromatin-remodeling complex) as well as activated β-catenin mutations are associated with HCC initiation and progression [59]. In addition, epigenetic mechanisms play an important role during the development and progression of HCC, and numerous studies have identified a large number of genes and pathways that are subject to epigenetic dysregulation [60]. Global DNA hypomethylation, histone modifications, promoter methylation, aberrant expression of noncoding RNAs, and dysregulated expression of epigenetic regulatory genes such as EZH2 are the best-known epigenetic abnormalities [61]. As mentioned above, alcohol metabolism alters the ratio of NAD+ to NADH and promotes the formation of ROS and acetate, all of which impact epigenetic regulatory mechanisms. Furthermore, the activities of enzymes involved in epigenetic modifications, such as DNA and histone methylation and histone acetylation, are influenced by the levels of metabolites such as NAD+, adenosine triphosphate (ATP), and S-adenosylmethionine (SAM). Chronic alcohol consumption leads to significant reductions in SAM levels, thereby contributing to DNA hypomethylation. These epigenetic changes are discussed in detail elsewhere [62] and are briefly mentioned below.
Epigenetic Effects
Since only about 10 % of heavy drinkers develop cirrhosis, the complex biological processes underlying states of health and disease could be determined by interactions between many genes and the external environment, and are likely to be driven by both genetic defects and by modifications that affect the transcriptional capacity of these genes (epigenetics). Epigenetic changes (e.g., DNA methylation or histone modification) affect gene expression directly or through the way DNA is packaged into chromatin, thus altering accessibility to transcription factors. In general, methylation of DNA represses gene expression by changing the chromatin structure or by interfering with the binding of some transcription factors to the promoter. DNA methylation has been shown to play a critical role in many cellular and biological processes, including cancer, aging, development, and the maintenance and differentiation of stem cells [63]. The successful establishment and maintenance of transcriptional profiles depend on the interplay between epigenetic modifications, interacting proteins, noncoding RNAs, and inter- and intrachromosomal interactions. Perturbation of any one of these regulatory elements may have profound consequences on the liver.
Chronic alcohol consumption has been shown to affect epigenetic regulation of gene expression involving DNA methylation, histone modification, and RNA-mediated gene silencing, thus modulating the expression of many genes. Alcohol-induced changes in gene expression may affect various biochemical and signaling pathways influencing the function of cells and organs, leading to liver disease and even cancer. Understanding alcohol-induced epigenetic regulation will provide mechanistic insights, diagnostic biomarkers, and therapeutic targets for alcohol-related liver injury.
Alcohol and DNA Methylation
DNA methylation tags cytosine, one of the four chemical bases that make up the genetic code, with a methyl group by transferring a methyl group from SAM onto the cytosine residue, which protrudes into the major groove of the DNA. Although acetaldehyde, the first metabolite of alcohol, can form DNA adducts and may cause sequence alteration of DNA, most of the short or long-lasting effects of alcohol may be independent of DNA sequence changes. Recent studies have shown that epigenetic regulation of gene expression is an important mechanism for alcohol’s action in the cell and alcohol-induced liver damage [38].
Alcohol-induced epigenetic changes may also affect stem cell differentiation and liver repair and regeneration. These processes are characterized by rapid, well-synchronized patterns of gene expression in which the status of DNA methylation shifts dramatically, involving both loss of methylation and de novo methylation. Many epigenetic changes during stem cell differentiation involve genes known to function in cell cycle, growth, apoptosis, and oxidative stress, all of which play a critical role in alcohol-induced liver damage and cancer. Evidently, purely sequence-based genetic or genomic approaches to study gene regulation are not sufficient to explain alcohol-related HCC.
Effects of Alcohol on the Availability and Transfer of Methyl Groups
Chronic alcohol consumption is associated with abnormal methionine metabolism, increased plasma homocysteine level, decreased level of SAM, and folate deficiency [5, 64]. SAM, the major methyl donor for DNA methylation, is primarily generated in the liver from l-methionine and ATP by methionine adenosyltransferase (MAT), which is encoded by two genes, MAT1A and MAT2A. MAT1A encodes the isoenzymes MATI and MATIII, whereas MAT2A gene encodes the isoenzyme MATII. MATI and MATIII are primarily responsible for maintaining high intracellular SAM levels in adult liver, while MATII is predominantly active in fetal and regenerating liver tissues.
Alcohol impairs the transfer of methyl groups to the cytosine residues of DNA by reducing the levels and activity of DNA methyltransferases (DNMT) , resulting in DNA hypomethylation. The alcohol metabolite acetaldehyde can also inhibit DNMT activity. Studies have shown that livers of MAT1A knockout mice had SAM deficiency and increased expression of genes involved in proliferation and consequently developed hepatomegaly, fatty liver, and eventually HCC. They also regenerated abnormally after partial hepatectomy and were more sensitive to developing steatosis in response to a methionine- and choline-deficient diet [65]. It has been reported that during hepatocarcinogenesis or hepatectomy, MAT1A itself is severely downregulated due to the hypermethylation of its promoter [5]. These observations suggest that alcohol may contribute to HCC development via its inhibition of MATI and MATIII activity as well as reducing SAM levels [66].
In addition to its effects on MAT and SAM synthesis, alcohol also inhibits a number of methyl group transfer-related enzymes such as methionine synthase and cystathionine-β-synthase. The latter removes homocysteine through trans-sulfuration to cystathionine, which is used to generate the antioxidant glutathione (GSH). The net result is a decreased level of GSH, leading to increased oxidative stress, also contributing to liver damage. Excessive alcohol intake can decrease the GSH level by inhibiting hepatic GSH synthesis and the enzymatic activities involved in GSH-related peroxide detoxification such as GSH peroxidase and glutathione S-transferase (GST), thus increasing the susceptibility of the liver to oxidative injury [67].
Another major site of alcohol’s actions on methyl group transfer is the folate metabolism cycle. Chronic alcohol consumption causes malabsorption of folates and increases their renal excretion resulting in a significant decrease in hepatic folate content [68]. In addition, it inhibits methionine synthase which transfers a methyl group from 5-methyl tetrahydrofolate to homocysteine to form methionine.
Although it is clear that chronic alcohol ingestion alters availability and transfer of methyl groups, its impact on DNA methylation appears to be complex. While DNA methylation in the promoter regions of class I ADH genes is elevated in an alcohol-treated human hepatoma cell line [69], the expression of DNA methyl transferase (DNMT-3b) is decreased in alcoholic patients [70]. The effect of alcohol on DNA methylation may depend on genomic context, cell type, and target organ.
Histone Modification
Chromatin structure is dynamically regulated to selectively facilitate the expression of some genes while maintaining others in a quiescent state and to allow for DNA repair and replication. Genes within highly condensed “heterochromatin” regions are generally silenced, whereas uncondensed “euchromatin” is permissive for gene expression. The various states of chromatin are largely attributable to the posttranslational modification of histones. These occur primarily on the N-terminal histone “tails” that protrude from the nucleosome structure and include mono-, di-, or tri-lysine methylation, lysine acetylation, lysine ubiquitination, arginine methylation (mono- or di-), and serine or threonine phosphorylation, among others.
Histone acetylation is associated with loosely packed chromatin and actively transcribed genes. As depicted in Fig. 13.3, histone acetylation is determined by the opposing activities of histone acetyltransferases (HATs) and deacetylases (HDACs). Histone methylation results from the action of histone methyltransferases (HMTs) and is reversed by histone demethylases. Enzymes involved in histone acetylation are relatively few in number and promiscuous in terms of which lysines they modify. In contrast, HMTs and histone demethylases are typically specific for a single H3 or H4 residue and, consequently, are more numerous [71].
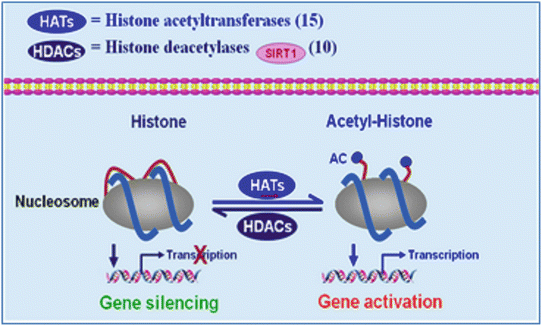
Fig. 13.3
Histone acetyltransferases (HATs) and deacetylases (HDACs) influence the acetylation, transcription, and condensation of chromatin
Alcohol and Histone Modification
Emerging evidence points to the potential of alcohol to exert its health effects by altering the state of chromatin. Acute alcohol administration to rats was shown to increase H3K9 acetylation in selected tissues, including liver, but not in others, indicating that epigenetic effects of alcohol will likely vary by tissue [72]. Consistent with these in vivo findings, in vitro studies found that alcohol also promotes the acetylation of H3K9 in primary hepatocyte cultures without affecting the acetylation status of other H3 lysines, including K14, K18, K23, or K27 [73]. In addition, this specific effect on H3K9 was associated with increased HAT activity by the acetate derived from alcohol metabolism. Accompanying this increase in H3K9 acetylation, there was an overall reduction of methylated H3K9 and a concomitant increase in methylated H3K4 in alcohol-treated hepatocyte cultures [74]. At the individual gene level, genes whose promoters exhibited this predominant pattern were found to be transcriptionally active, while those exhibiting the inverse pattern (i.e., increased H3K9me, decreased H3K4me) were silenced. Together, these findings highlight the potential for alcohol to alter patterns of histone modification and the expression of associated genes, raising the possibility that oncogenes and/or tumor suppressors might be among the affected genes and represent a mechanism by which alcohol contributes to HCC.
The decrease in NAD+/NADH ratio due to alcohol metabolism has the potential to diminish the activity of NAD+-dependent enzymes, including the SIRT family of histone deacetylases (HDACs). Indeed, inhibition of hepatic SIRT1 activity by alcohol was associated with an increase in the acetylated active nuclear form of SREBP-1c in the livers of alcohol-fed mice leading to impairment of lipid metabolism [75].
MicroRNAs, Cancer, and Alcohol
MicroRNAs (miRNAs) often regulate the expression of cancer pathway components, including oncogenes and tumor suppressors. While miRNAs are often globally downregulated in human tumors [76], some miRNAs are frequently dysregulated in many types of cancer. The degree to which the expression of some miRNAs is altered is correlated with clinical or pathologic indicators of malignancy, offering opportunities for early detection and insights into early pathogenesis [77].
HCC is of particular relevance to alcohol and has been the subject of several miRNA profiling studies. Murakami et al. [78] identified three miRNAs that were overexpressed in HCC (miR-224, miR-18, and pre-miR-18) and five others that were under-expressed (miR-199a, miR-199a*, miR-200a, miR-125a, miR-195). Furthermore, they demonstrated the effectiveness of a signature, based on these changes, in distinguishing HCC and non-HCC cases and identified three miRNAs (miR-92, miR-20, and miR-18) whose expression was inversely correlated with the degree of HCC differentiation. Another study defined miRNA changes that allowed differentiation of HCC (increased miR-21, miR-10b, and miR-222) from benign hepatocellular adenomas (decreased miR-200c and miR-203) [79]. Importantly, this study also revealed specific miRNA markers of alcohol-related HCC (decreased miR-126) and HCC associated with hepatitis B viral exposure (increased miR-96). The molecular pathways regulated by miRNA in HCC are detailed in a review by Milazzo and colleagues [80].
Effect of Alcohol on miRNA Expression and Function
Given the breadth of processes that miRNAs are known to regulate, it is reasonable to expect that they will play significant roles in mediating the effects of alcohol, including cancer. Several recent reports have identified miRNAs whose levels are altered by alcohol and that mediate alcohol’s ability to promote gut leakiness [81]. Chronic alcohol consumption increased miR-21 expression during liver regeneration in ethanol-fed rats [82] and enhanced miR-155 in macrophages via NF-6B, which contributed to the elevation in TNF-α production [83]. In addition, chronic alcohol feeding resulted in significant alteration in several miRNAs that regulate hepatic metabolism (miR-34a, miR-103, miR-107, and miR-122) [84], as well as downregulation of miR-199, which may contribute to HIF-1α augmentation [85]. In addition, large-scale miRNA screens indicate that alcohol alters the expression of 2–3 % of miRNAs (miR-320, miR-486, miR-705, and miR-1224 and a decreased expression for miR-27b, miR-214, miR-199a-3p, miR-182, miR-183, miR-200a, and miR-322) in murine models of alcohol-induced steatohepatitis [86]. These changes in miRNAs and their significance were reviewed elsewhere [87].
Circadian Rhythm Perturbation
The heterodimer of transcription factors CLOCK and BMAL1, which activates transcription of the period (Per) and cryptochrome (Cry) genes, comprises the centerpiece of the mammalian circadian network [88]. Their products PER and CRY interact to form the PER/CRY complex which translocates into the nucleus to inhibit CLOCK/BMAL1 transactivation, which in turn results in the repression of the Per and Cry genes. Release of PER/CRY complex through proteasome degradation can relieve repression and start the negative feedback loop again.
Despite the fact that there is no direct evidence connecting circadian rhythm disturbance to alcohol-induced HCC, dysfunctions of circadian rhythms are involved in many diseases that are known to be modulated by alcohol. The hypothesis that disturbance in circadian rhythms by alcohol may be involved in cancer is supported by the following research:
Disturbance in circadian rhythm genes expression is a common feature in certain types of cancer, including HCC [89]. Many studies have suggested indirectly that circadian rhythms may play an important role in hepatocarcinogenesis. Key clock genes have been found to be disrupted in HCC patients [90], demonstrating that HCC impacts the orchestrated circadian rhythm of liver cells. In addition, a long noncoding RNA (lncRNA), highly upregulated in liver cancer (HULC), has been reported to contribute to the perturbations in circadian rhythm of hepatoma cells [91]. Furthermore, a single functional polymorphism of one SNP rs2640908 in PER3 gene was significantly associated with overall survival of HCC patients [92].
Liver metabolism can be greatly affected by circadian rhythms and changes in feeding status. A large number of metabolic enzymes, such as CYP2E1, CYP3A4, CYP3A11, ADH, and ALDH—many of which are involved in alcohol metabolism—are regulated by the circadian clock [93]. Many other important metabolic pathways such as glycolysis, fatty-acid metabolism, cholesterol biosynthesis, and xenobiotic and intermediate metabolism are also under circadian regulation [94]. The rate-limiting steps of these metabolic pathways are often the target sites of circadian control. Because of their important roles in metabolism, mutations in the clock genes often cause metabolic disorders. For example, a mutation of the Clock gene caused mice to be hyperphagic and obese, exhibiting hyperlipidemia, hepatic steatosis, hyperglycemia, and hypoinsulinemia [95]. Conversely, circadian clocks can be entrained by various metabolism-related external cues, such as food intake and alcohol consumption. A high-fat diet in mice can change the expression of clock genes and clock-controlled genes [96], delay the circadian expression of adiponectin signaling components, and inhibit AMPK expression in mouse liver [97]. Furthermore, some nuclear receptors, such as PPARα, PPARγ, glucocorticoid receptor, RARα, and RXRα, have been directly connected to the key components of the circadian system [98] and have been also implicated in alcohol-induced tissue injury.
Some biological pathways closely related to alcohol’s actions are involved in, or affected by, circadian rhythms. The redox state of cells plays an important role in the function of the circadian rhythm. The presence of NADH or NADPH promotes the binding of the heterodimeric clock transcription factor complexes to DNA [99]. In addition, studies suggest that the histone deacetylase SIRT1 may be involved in the integration of circadian and metabolic transcription networks. It has been shown that SIRT1 interacts directly with CLOCK and deacetylates BMAL1 and PER2. Malondialdehyde, a marker of oxidative stress affected by alcohol, is found to exhibit circadian patterns of expression in mice liver [100]. The retinoic acid receptors RXR and RAR can interact with CLOCK and NPAS2 (a homolog of CLOCK), and these interactions can be increased 15-fold by retinoic acid [101]. Other consideration is that circadian rhythm disturbances lead to immunodeficiency [102], suppression of natural killer cell activity, and alteration in the T-helper 1/T-helper 2 cytokine balance, resulting in decreases in cellular immunity and tumor immune surveillance [103], all of which are affected by alcohol.
Lipopolysaccharide (LPS) , which is increased in blood after alcohol consumption, suppresses clock genes, suggesting that circadian rhythms play an important role in response to systemic inflammatory stimulation [104].
The relative contributions of disrupted circadian rhythm and circadian genes to cancer risk may be informative as to the pathogenesis of various cancers and new treatment interventions.
Immune Modification
The first line of host defense against HCC is innate immunity, including natural killer (NK) cells and T lymphocytes. The involvement of the immune system in HCC carcinogenesis has been previously proposed in clinical studies, where the percentage and absolute number of NK cells were decreased significantly during the development and progression of HCC [105]. Effective adaptive immune response depends on the antigen-specific activation of T and B cells. Increased activity of helper T cells, which promote inflammation, is associated with HCC [106], and chronic inflammation has been implicated in the development of liver cancer in humans [107]. While the immunoglobulin protein family (CD28 and cytotoxic T-lymphocyte antigen-4) plays important roles in the control of T-cell responses against infection and cancer, tumors seem to have exploited these pathways to evade immune surveillance. Activation and proliferation of cytotoxic T lymphocytes is suppressed in individuals with HCC [108].
Recent studies suggested a role of the immune system in constitutional susceptibility to HCC. A genome-wide association study (GWAS) , focusing on HCC, revealed that constitutional genetic variations are risk factors for HCC [109]. Three susceptibility loci have been strongly associated with HCC including the class II MHC complex, whose protein products present antigen to T-cell receptors and mediate immune surveillance (rs9267673, rs2647073, and rs3997872) resulting in an ineffective T-cell response. MHC class II molecules present antigen to CD4. Thus, genes involved in the immune response play a critical role in the development of HCC. Since only a subset of liver cirrhosis patients develop HCC, the transition from cirrhosis to HCC has been attributed to two SNPs whose allele frequencies differ significantly between HCC and cirrhosis (one lies in the PTEN homolog TPTE2, and the second variant lies within an intron of TPTE2, which encodes a homolog of the PTEN tumor suppressor protein [110]). Multiple SNP analysis showed that “antigen processing and presentation” emerged as the pathway with the strongest association with HCC. Thus, the T-cell repertoire of each individual plays a critical role in HCC susceptibility and that biological processes affecting T-cell maturation or immune surveillance may represent important etiologic mechanisms for the development of HCC in humans.
Related posts:
 Nonalcoholic Fatty Liver Disease in Children
Diagnostic Considerations and Clinical End Points for Nonalcoholic Steatohepatitis
Hepatic and Extrahepatic Malignancies in NAFLD
Nonalcoholic Fatty Liver Disease in Children
Diagnostic Considerations and Clinical End Points for Nonalcoholic Steatohepatitis
Hepatic and Extrahepatic Malignancies in NAFLD
 Clinical Features, Disease Modifiers, and Natural History of Alcoholic Liver Disease
Clinical Features, Disease Modifiers, and Natural History of Alcoholic Liver Disease
 Animal Models of Alcoholic Liver Disease
Animal Models of Alcoholic Liver Disease
 Epidemiology and Risk Factors for Alcoholic Liver Disease
Epidemiology and Risk Factors for Alcoholic Liver Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


