Fig. 7.1
NAFLD and AFLD share similar underlying pathogenic mechanisms and a common underlying genetic component. The cartoon depicts the disease progression of NAFLD and AFLD from normal liver to steatohepatitis and advanced disease with or without liver fibrosis. Liver triglyceride accumulation is a common underlying process in both diseases, and the transition from simple steatosis to progressive disease involves both NAFLD and AFLD oxidative stress, inflammation, mitochondrial damage, and fibrogenesis. The genes reported to be involved in these disease pathogenic pathways are highlighted in light blue. PNPLA3, patatin-like phospholipase domain-containing protein 3; CYP2E1, cytochrome P450, family 1, subfamily A, polypeptide 2; PPARA, peroxisome proliferator-activated receptor alpha; PPARGC1A, peroxisome proliferator-activated receptor gamma, coactivator 1 alpha; PTGS2, prostaglandin-endoperoxide synthase 2 (prostaglandin G/H synthase and cyclooxygenase); PDGFA, platelet-derived growth factor alpha polypeptide; LEPR, leptin receptor; APOE, apolipoprotein E; ADIPOR2/1, adiponectin receptor 2/1; ABCB11, ATP-binding cassette, subfamily B (MDR/TAP), member 11; TNFa, tumor necrosis factor a; SOD2, superoxide dismutase 2, mitochondrial; TLR4, toll-like receptor 4; IL-6, interleukin 6 (interferon, beta 2); IL-10, interleukin 10; IL-1R, interleukin 1 receptor; STAT3, signal transducer and activator of transcription 3 (acute-phase response factor); NFKB, nuclear factor of kappa light polypeptide gene enhancer in B cells; CD14, monocyte differentiation antigen CD14; GSTM1, glutathione S-transferase mu 1; KLF6, Kruppel-like factor 6; AT1R, angiotensin II receptor, type 1; MTTP, microsomal triglyceride transfer protein; GCKR, glucokinase (hexokinase 4) regulator; TGFB1, transforming growth factor, beta 1; ADH2/3, alcohol dehydrogenase 2/3; ALDH, aldehyde dehydrogenase 2 family (mitochondrial)
The knowledge regarding the pathogenesis NAFLD and its genetic component has significantly increased in the last 10 years when clinicians, gastroenterologists, and hepatologists started to realize the harmful health consequences of the obesity and type 2 diabetes pandemic that affected Western countries initially but now has expanded globally [7].
During the 1980s, Ludwig regarded nonalcoholic steatohepatitis (NASH) as a poorly understood and hitherto unnamed liver disease that histologically mimics alcoholic hepatitis and that also may progress to cirrhosis [8]. Nevertheless, “fatty liver” is not an emerging disease for hepatologists; on the contrary, fatty liver has been a “hub” of intense and fructiferous research for over 30 years. For example, in 1961, Charles Lieber observed that fatty infiltration of the liver was a common finding in alcoholic patients [9]. Interestingly, Lieber showed that unlike adipose tissue, ethanol is oxidized by alcohol dehydrogenase (ADH) in the liver, and he suggested that the excess ethanol in the liver leads to fatty acid biosynthesis [9] (Fig. 7.2). Four years later, Lieber performed studies in patients who had a history of alcohol consumption by doing liver biopsies during and after alcohol ingestion [10]; remarkably, Lieber’s observations showed that the steatosis seemed to depend on both the dose and the duration of ethanol intake. It was not possible to establish a threshold for the production of fatty liver by ethanol because some individuals had liver damage after exposure to high amounts of alcohol while others did not [10].
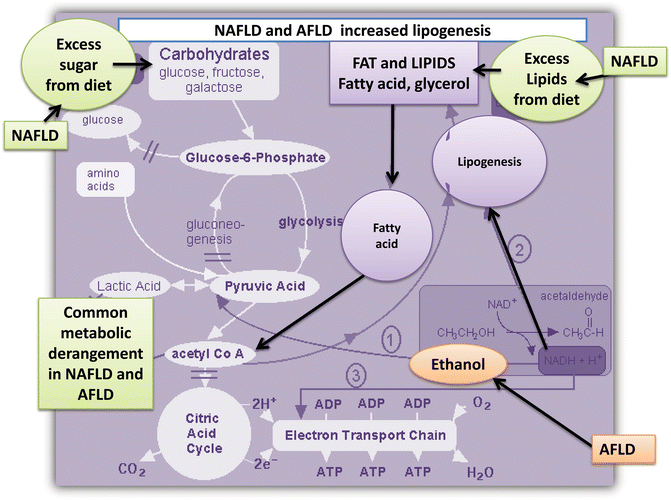
Fig. 7.2
NAFLD and AFLD are associated with increased lipogenesis. The cartoon illustrates the metabolic pathway associated with increased lipogenesis in NAFLD (increased energy from diet) and AFLD (metabolism of ethanol that feeds the fatty acid metabolism cycle by converting to acetaldehyde)
This observation can be translated into a modern concept of the etiology of human diseases. This concept assumes that a genetic component that illustrates the interindividual differences in response to similar insults can explain the disease susceptibility of complex diseases.
On the other hand, NAFLD and AFLD share not only the degree of histological changes, including the presence of lobular inflammation, morphological changes in liver mitochondria, perivenular and perisinusoidal fibrosis, and even hepatocellular ballooning [11, 12], but also, as we have shown recently, they seem to share similar underlying molecular disease mechanisms. From a genetic point of view, they are difficult to distinguish from each other [4]. More importantly, hepatic fat accumulation in NAFLD and AFLD is connected by a common metabolic pathway (Fig. 7.2) that involves enzymatic reactions that impact on the citric cycle [13–15]. Furthermore, as ethanol increases the rate of synthesis of fat [16], an increasing number of lipogenic enzymes show increased transcriptional activity in both NAFDL and AFLD [16].
In this chapter, we review the current knowledge and recent insights regarding the genetic basis of NAFLD and AFLD in an integrative approach to understand the role of the genetic component in the susceptibility of abnormal liver fat accumulation and the progression of liver disease.
The Genetic Component of NAFLD and AFLD and the Role of PNPLA3 Gene on Liver Fat Accumulation
The genetic component of polygenic human diseases can be studied by two main approaches: candidate gene association studies, which are, in general, hypothesis driven, and genome-wide association studies (GWAS). Although advances in genotyping technology and information generated by GWAS have expanded our knowledge about gene variants associated with complex diseases, including NAFLD and AFLD, many important questions remain unanswered. For example, single-nucleotide polymorphisms (SNPs) identified by GWAS are not necessarily involved in the disease biology because they do may represent the causal variant.
A growing body of evidence indicates that NAFLD [5] and AFLD [6] develop from a complex process in which many factors, including genetic susceptibility and environmental insults, are involved. Data from the first GWAS on NAFLD [17] have significantly contributed to our knowledge of the genetic component of fatty liver, as it stemmed the search for replication of its findings in patients with a myriad of liver diseases, including AFLD [18].
Nowadays, the nonsynonymous SNP, rs738409 C/G, of PNPLA3 (patatin-like phospholipase domain-containing protein 3, also known as adiponutrin or calcium-independent phospholipase A2-epsilon), encoding an amino acid substitution I148M, is regarded as the major genetic component of hepatic triglyceride accumulation and fatty liver disease, including NAFLD [19] and AFLD [20]. In fact, the risk effect of the rs738409 on developing fatty liver in the context of NAFLD is perhaps one of the strongest ever reported for a common variant modifying the genetic susceptibility for complex diseases (5.3 % of the total variance) [19].
In addition, the rs738409 is not only significantly associated with the accumulation of fat in the liver but also with the histologic disease severity and progression of NAFLD (OR 1.88 per G allele; 95 % CI = 1.03–3.43; p < 0.04) [19] and the development of cirrhosis in AFLD (OR = 2.08; 95 % CI = 1.15–3.77; p = 0.02) [21].
Indeed, the rs738409 variant not only modifies the biology of NAFLD but also has a considerable impact on the genetic susceptibility to alcoholic liver disease (ALD) [20–22] and hepatitis C- [23] and hepatitis B-induced fatty liver, and it has also been associated with hepatocellular carcinoma occurrence among patients with cirrhosis [24–26], indicating that these diverse liver diseases may share common pathophysiological pathways associated with accumulation of the fat in the liver.
PNPLA3 is a multifunctional enzyme with both triacylglycerol (TAG) lipase and acylglycerol O-acyltransferase activity that participate in TAG hydrolysis and the acyl-CoA-independent transacylation of acylglycerols [27]. Interestingly, nutritional factors, including oleic acid, C18:2 fatty acid, palmitic acid, glucose, insulin, and lactone, induce PNPLA3 [27]. In addition, the promoter activity of PNPLA3 is upregulated by glucose concentrations in a dose-dependent manner [28].
The basic function of PNPLA3 was explored in adipose tissue initially and can be summarized as follows: (1) it is strongly associated with membranes and with lipid droplets; (2) it has triacylglycerol lipase activity that mediates triacylglycerol hydrolysis in adipocytes; (3) it may be involved in the balance of energy usage/storage in adipocytes; (4) it participates in triacylglycerol hydrolysis and the acyl-CoA-independent transacylation of acylglycerols, thereby facilitating energy mobilization and storage in adipocytes [29]; (5) it has lipid hydrolase with an unusual folding topology that differs from classical lipases; (6) its function may be related to some regulatory aspect of the pathway of lipogenesis or lipolysis; and (7) it plays a role in the hydrolysis of glycerolipids [30]. Figure 7.3 depicts the KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway of glycerolipid metabolism , in which PNPLA3 involvement is highlighted. Nevertheless, the role of PNPLA3 in the liver seems to be broader than adipose tissue, involving hepatocyte triacylglycerol remodeling [18, 30] and global metabolic changes [31].
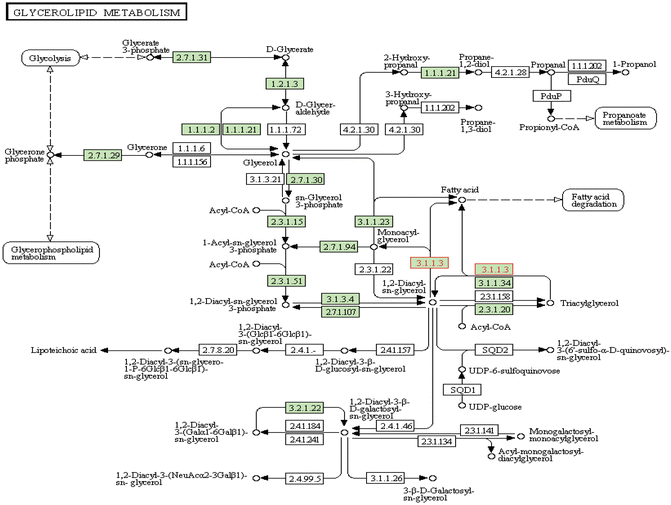
Fig. 7.3
The KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway of glycerolipid metabolism. The pink highlights the 3.1.1.3 enzymatic reactions involved in acylglycerol degradation, in which PNPLA3 is involved
Whether the rs738409 variant is associated with a gain or loss of function has been a matter of debate [18]. For example, Kumari et al. reported that PNPLA3 promotes cellular lipid synthesis by converting lysophosphatidic acid into phosphatidic acid; hence, the I148M substitution promotes hepatic lipid synthesis due to a gain of function [32]. Conversely, in vitro examination of PNPLA3 enzymatic activity showed that the wild-type protein shows a predominant lipase activity with mild lysophosphatidic acid acyl transferase activity while the I148M mutation results in a loss of function of both these activities [33]. Likewise, more recently, Smagris and coworkers explored the effect of introducing a methionine codon at position 148 of the mouse PNPLA3 gene; when mice were challenged with a high-sucrose diet, their liver fat levels increased two to threefold compared with wild-type littermates [34]. The authors also showed that the catalytically inactive PNPLA3 on the surfaces of lipid droplets was associated with liver fat accumulation [34].
Finally, a recent study uncovered the functional role of PNPLA3 on liver metabolism by performing high-throughput metabolic profiling of PNPLA3 siRNA silencing and overexpression of wild-type and mutant Ile148Met variants (isoleucine/methionine substitution at codon 148) in Huh-7 cells [31]. Of note, the silencing of PNPLA3 was associated with a global perturbation of Huh-7 hepatoma cells that resembled a catabolic response associated with protein breakdown; a significant decrease in amino- and gamma-glutamyl amino acids and dipeptides and a significant increase in cysteine sulfinic acid, myo-inositol, lysolipids, sphingolipids, and polyunsaturated fatty acids were observed [31]. On the other hand, overexpression of the PNPLA3 Met148 variant mirrored many of the metabolic changes observed during gene silencing but in the opposing direction. Interestingly, overexpression of the PNPLA3 Met148 variant was associated with a 1.75-fold increase in lactic acid in comparison with the empty vector, suggesting a shift to anaerobic metabolism and mitochondrial dysfunction. Together, these results suggest a critical role of PNPLA3 in the modulation of liver metabolism beyond its classical participation in triacylglycerol remodeling [31] and could explain the role of PNPLA3 in disease progression.
GWAS on NAFLD and the Genetic Risk of Disease Progression
The first GWAS on NAFLD was a genome-wide survey of nonsynonymous sequence variations encompassing 9229 SNPs in a multiethnic population-based study [17]. The use of the GWAS strategy in the search for the genetic component of NAFLD was followed by other reports that included different populations, study designs, sample sizes, and approaches for the characterization of the main liver phenotype, for example, female adults with NAFLD diagnosed by liver biopsy [35], exploration of the heritability of hepatic steatosis at the population level with computed tomography [36], a combined approach of CT and alanine aminotransferase (ALT) levels as a surrogate of disease severity [37], exploration of the genetic risk in Asian-descent patients [38, 39], and liver fat content in extremely obese individuals [40].
It is also important to highlight that the coverage of SNPs by the abovementioned GWAS was not uniform in terms of the explored variants. In addition, it varied from a GWAS analysis of 12,138 nonsynonymous sequence variations from dbSNP and the Perlegen SNP database [17] to commercial platforms, such as HumanCNV370-Quadv3 BeadChip (coverage: 373,397 SNPs) [35] or Illumina Human 610-Quad BeadChip (coverage: 484,751 SNPs), meta-analysis, and GWAS association data of large consortiums that used the Affymetrix 6.0 or Illumina platform [36] and imputed SNPs [37].
Finally, the GWAS strategy was also used to explore the genetic locus that influenced liver enzyme levels in the population, including ALT [41, 42]. Figure 7.4 depicts a summary of the latest GWAS on NAFLD and ALT levels. Of note, these GWAS uncovered loci whose function is diverse but interesting in the context of NAFLD. For instance, PPP1R3B (protein phosphatase 1, regulatory subunit 3B) is associated with the regulation of glycogen synthesis in liver or skeletal muscle; FDFT1 (farnesyl-diphosphate farnesyltransferase) is involved in cholesterol biosynthesis; ERLIN1 (ER lipid raft-associated 1) mediates the endoplasmic reticulum-associated degradation; LTBP3 (latent transforming growth factor beta) plays a structural role in the extracellular matrix; and PARVB (parvin beta) plays a role in cytoskeleton organization and cell adhesion. Conversely, certain loci, such as CPN1 (carboxypeptidase N, polypeptide 1), NCAN (neurocan), and EFCAB4B (EF-hand calcium-binding domain 4B), whose biological function seems to be distant from the pathogenesis of NAFLD are still worth being explored from the mechanistic point of view to reveal their role in disease susceptibility. In fact, rs2228603 in NCAN locus is an interesting example of misinterpretation of casual variants in NAFLD-GWAS [36]. While no functional study was done on the abovementioned NCAN variant, it was speculated that rs2228603[T] allele is a risk factor for liver inflammation and fibrosis, suggesting that this locus is responsible for progression from steatosis to steatohepatitis [43]. Furthermore, despite the biological plausibility of a putative role of NCAN in NAFLD being hard to support as this gene codes for a chondroitin sulfate proteoglycan expressed primarily in nervous system, it was speculated that rs2228603 is associated with a “brain-liver axis” that, when deregulated, increases the risk for NAFLD [43]. Remarkably, recently findings from an exome-wide association study [44], followed by replication studies in different population around the world [45–49], have definitively showed that the causal variant in the multigene locus named NCAN/TM6SF2/CILP2/PBX4 is the nonsynonymous variant located in the TM6SF2 (transmembrane 6 superfamily member 2) gene, the rs58542926 encoding an amino acid substitution p.Glu167Lys (E167K). The initial study of Kozlitina et al. showed that rs58542926 was significantly associated with hepatic triglyceride content (HTGC) as measured by proton magnetic resonance spectroscopy (H-MRS). The authors showed that the effect of the rs58542926 on HTGC was independent of the effect mediated by the rs738409, obesity, insulin resistance as assessed by HOMA-IR, or alcohol intake [44].
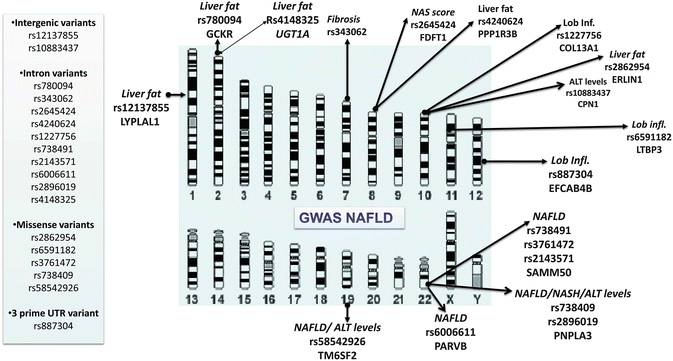
Fig. 7.4
GWAS on NAFLD : Summary representation of variants significantly associated with NAFLD, NASH, and plasma levels of alanine aminotransferase (ALT). The illustration depicts the chromosome localization of significantly associated SNPs according to the main NAFLD phenotypes. LYPLAL1, lysophospholipase-like 1; GCKR, glucokinase (hexokinase 4) regulator; COL13A1, collagen, type XIII, alpha 1; PPP1R3B, protein phosphatase 1, regulatory subunit 3B; ERLIN1, ER lipid raft-associated 1; EFCAB4B, EF-hand calcium-binding domain 4B; TM6SF2, transmembrane 6 superfamily member 2; PARVB, parvin beta; PNPLA3, patatin-like phospholipase domain-containing protein 3; SAMM50, sorting and assembly machinery component 50 homologue (S. cerevisiae); LTBP3, latent transforming growth factor beta-binding protein 3; FDFT1, farnesyl-diphosphate farnesyltransferase 1; CPN1, carboxypeptidase N, polypeptide 1; UGT1A, glucuronosyltransferase 1 family
Subsequent studies showed conflicting results as some [45, 47–49] but not all studies [46, 50] showed a significant association with fatty liver or histological steatosis, and the association with liver fibrosis reported by one large study [46] remains to be confirmed as most of the studies showed that the association does not resist adjustment by NASH [45] or was not significant [47, 50]. In fact, a recent study on chronic hepatitis C showed that while TM6SF2-E167K variant is an independent predictor of liver, no difference in necroinflammatory or fibrosis scores was found among carriers and noncarriers of the risk allele [51]. Finally, functional studies on TM6SF2 gene demonstrated that this locus and the mentioned variant are relevant on NAFLD disease biology. For instance, Kozlitina et al. showed in vitro that murine hepatoma cells expressing the Lys167-TM6SF2 (E variant) protein have reduced expression levels compared with the wild type [44], and Mahdessian demonstrated that TM6SF2 is localized in the endoplasmic reticulum and the ER-Golgi intermediate compartment of human liver cells [52]. Moreover, to understand whether the rs58542926 genotypes have any effect on human NAFLD, our group explored the level of liver TM6SF2 expression in subjects with NAFLD at different stages of disease severity [47]. Interestingly, we observed that TM6SF2 protein expression was significantly reduced in the liver of patients with NAFLD; reduced expression of liver TM6SF2 was associated with a high degree of steatosis and NAS score. In addition, we noted that liver TM6SF2 immunoreactivity was reduced in carriers of the NAFLD-risk T allele (Lys167); allelic-specific expression analysis of cDNA isolated from the liver tissue confirmed that expression levels of rs58542926-T are about 56 % of that of the C allele. Taken together, these findings suggest that the TM6SF2-NAFLD-risk T allele is associated with decreased gene and protein expression in the liver of affected patients [47].
Of note, as we initially noticed, the TM6SF2-rs58542926 presents a clinical paradox as the C (Glu167) allele is associated consistently with increased cardiovascular risk by increasing circulating LDL cholesterol [53] and replicated by others [44, 45, 47] but the other T allele (Lys167) is associated putatively with NAFLD and NASH. Nevertheless, it is important to note that rs58542926 is a low-frequency variant with a rather modest putative protective and risk effect on CVD and NAFLD, respectively. This observation supports the concept that other than “common-frequent” variants may contribute to the heritability of NAFLD and may also explain, at least in part, the missing heritability problem.
Finally, the association of the rs58542926 and the level of serum transaminases remain to be elucidated because the association was not replicated consistently in different cohorts as we recently summarized [47].
Evidence About the Heritability of NAFLD and Related Disease Phenotypes
The search of the genetic component of complex diseases is enriched by research studies that explore the hereditability of a given disease by performing familial aggregation studies. This kind of study is focused on determining whether the index cases (probands) have relatives that cluster similar phenotypes; hence, familiar aggregation studies are devoted primarily to seeking whether having relatives with disease increases one’s risk of that disease.
Struben and colleagues provided the first evidence on the heritability of NAFLD when they examined the familial pattern of cryptogenic cirrhosis by reviewing the family history of patients with NASH with and without cirrhosis or cryptogenic cirrhosis to assess how frequently their relatives were afflicted with these disorders [54]. The authors included 18 members of eight kindreds containing two or more afflicted members and observed that NASH coexisted within four kindreds; the pattern of afflicted patients included mother–daughter, sister–sister, sister–brother, father–daughter, and male–female cousins [54].
Schwimmer et al. performed a familial aggregation study of fatty liver in overweight children with and without NAFLD and demonstrated that fatty liver was present in 17 % of siblings and 37 % of parents of overweight children without NAFLD [55]. Abdelmalek et al. described familial aggregation of insulin resistance in first-degree relatives of patients with NAFLD [56], and Loomba reported similar findings in patients from the Nonalcoholic Steatohepatitis Clinical Research Network that explored the family history of type 2 diabetes in subjects with NASH and NAFLD and concluded that family history of diabetes, especially among nondiabetics, is associated with NASH and fibrosis in NAFLD [57].
Finally, an elegant study on the University of California at San Diego twin cohort with 362 twins showed that plasma gamma-glutamyl transpeptidase (GGT) is a heritable trait and that GGT shares significant genetic covariance with uric acid, insulin, HOMA-IR, triglycerides, and blood pressure [58].
Likewise, unpublished data from the “The Genetics of NAFLD in Twins Consortium” led by Rohit Loomba and colleagues demonstrated that while both hepatic steatosis and hepatic fibrosis are heritable traits, they appear to have distinct basis for their genetic susceptibility. Notably, genetic covariance assessment revealed a significant association between hepatic steatosis and BMI and hyperinsulinemia and between hepatic fibrosis and HbA1c (Rohit Loomba, unpublished personal communication).
Ethanol Metabolism and Genetic Variants Influencing Alcoholic Liver Disease
Once ingested, several enzymatic and nonenzymatic mechanisms in the liver and other organs like the stomach metabolize ethanol. The first step of ethanol metabolism is its oxidation into acetaldehyde, which is oxidized subsequently to acetate through several enzymatic reactions, including aldehyde dehydrogenase. The alcohol dehydrogenase family is composed of by at least seven genes and comprises at least five classes [59]; isozymes are distributed differentially in tissues, with most classes exhibiting the highest activity in liver. Alcohol dehydrogenase (ADH1), which consists of several homo- and heterodimers of alpha, beta, and gamma subunits, catalyzes the oxidation of alcohols to aldehydes; three genes encoding alpha, beta, and gamma subunits are organized tandemly in a genomic segment as a gene cluster. ADH1A is active in the liver in early fetal life but only weakly active in adult liver. Alcohol dehydrogenase 1B (ADH1B, class I) beta polypeptide is a protein that exhibits high activity for ethanol oxidation and plays a major role in ethanol catabolism. ADH1C is a paralog of ADH1A and B. ALDH2 (aldehyde dehydrogenase 2 family) is the second enzyme of the major oxidative pathway of alcohol metabolism, with two major liver isoforms of aldehyde dehydrogenase, cytosolic and mitochondrial. ADH4 (alcohol dehydrogenase 4 (class II), pi polypeptide) exhibits a high activity for oxidation of long-chain aliphatic alcohols and aromatic alcohols and is less sensitive to pyrazole.
Genetic polymorphisms in the ADH and ALDH families of genes are responsible for the high individual variability in ethanol metabolism, as gene variants determine the level of acetaldehyde accumulation after alcohol consumption [6, 60, 61]. Around 50 % of East Asian subjects carry an inactive ALDH2 gene and exhibit acetaldehyde accumulation after alcohol consumption. Interestingly, compared with wild-type mice, ethanol-fed ALDH2–/– mice had higher levels of malondialdehyde-acetaldehyde (MAA) adduct and greater hepatic inflammation, with higher hepatic interleukin (IL)-6 expression but lower levels of steatosis and serum alanine aminotransferase (ALT) [62].
Another important family of genes that mediates ethanol metabolism is the cytochrome P450 superfamily of enzymes that are monooxygenases, which catalyze many reactions involved in drug metabolism and the synthesis of cholesterol, steroids, and lipids. Cytochrome P450, family 2, subfamily E, polypeptide 1 (CYP2E1) localizes to the endoplasmic reticulum and is induced by ethanol; thus the enzyme metabolizes both endogenous substrates, such as ethanol, acetone, and acetal as well as exogenous substrates. A recent meta-analysis showed that CYP2E1 might be significantly associated with the development of steatosis, hepatitis, and fibrosis in patients with AFLD [63]. It is worth mentioning that there are no published GWAS in AFLD, though two efforts are ongoing—one is a multinational GWAS of alcoholic cirrhosis and another is a US-based GWAS of acute alcoholic hepatitis.
The Role of Epigenetic Mechanisms in the Development and Disease Progression of NAFLD and AFLD
Genetic factors other than DNA variation are likely to play an important role in the etiology of complex diseases, specifically epigenetic modifications. By definition, epigenetic factors, which are the most important as described below, are DNA methylation and covalent histone modifications typically at lysine (K) in their N terminal regions. These are modifiers of gene expression that without altering the DNA sequence itself, are capable of self replication through cell mitosis and even of being transmitted to the next organismal generation. Actually, the most interesting points about epigenetic modifications are that they are crucial during development and they are potentially modified and disrupted by environmental influences, mainly dietary and behavioral habits and therapeutic intervention.
Cytosine (C) methylation (5-methylcytosine) is a common epigenetic modification, where a C is adjacent to a guanine (G) nucleotide (CpG). In the whole mammalian genome, around 5 % of cytosines are methylated, typically outside of the so-called CpG islands (regions of typically 300–3000 bp in length with a high content of CpG and C/G %). Nearly 50–60 % of genes have CpG-rich islands in their 5′ untranslated regions near or in the promoters. During fetal development, as well as in adult life, normal somatic cells (and cancer cells) exhibit alterations in DNA methylation induced by environmental stimuli. As CpGs are paired with GC in the opposite strand, methylation in one strand is mirrored by methylation in the other. During replication, methylation in the parent strands directs methylation in the newly replicated DNA by recruiting DNA methyltransferases. Subsequently, stable transfer of gene methylation patterns to progeny lines is accomplished. CpG methylation is thought to constrain expansive regions of the genome by silencing repetitive sequences or repressing promoters by recruiting methyl-CpG-binding proteins (the MBD protein family). Although methylation is associated frequently but not always, with repressed promoters, transcriptional repression via histone methylation and/or deacetylation precedes DNA methylation. Table 7.1 gives a short overview of the main features of epigenetic modifications.
Table 7.1
A brief overview of epigenetic modifications and their effects on gene regulation
Epigenetic modification | Main outcome and effect |
|---|---|
1-DNA methylation | • Modulates gene transcription • Methylation of CpG islands of promoter region is mostly associated with silencing of gene expression • Methylation of CpG dinucleotides located in the gene coding sequence is associated weakly with gene silencing but the opposite |
Methylation is rare in CpG-rich regions or CpG islands | |
Regions are usually longer than 500 bp | |
GC base content > 55 % | |
Located in the promoter regions and at the end of the 5′ region. Types of promoters: rich and poor in CpG islands | |
Methylated sites are distributed globally on about 80 % of CpGs | |
Enzymes involved in this process: DNA methyltransferases (DNMTs) DNMT1, DNMT3a, DNMT3b, DNMT2, and accessory proteins, such as DNMT3L | |
Stable in somatic cells but modifiable by environmental factors | |
Methylation levels may show interindividual heterogeneity | |
Different tissues are able to show local differences in DNA methylation | |
Non-CpG methylation | • Potentially inducible by environmental factors |
Location in the promoter remains controversial | |
The functional significance of non-CpG methylation in early development is uncertain | |
May be modulated by DNMT3 activity | |
Can occur in CpA, CpC, and CpT nucleotides situated in the DNA of embryonic stem cells and episomal DNA | |
2-Histone posttranslational modifications | • Implicated in the de novo methylation of DNA • Histone acetylation: associated with more open chromatin and transcriptional activation • Histone hypoacetylation: Associated with condensed chromatin structure and repression of gene transcription |
Modifications: – Acetylation, methylation, ubiquitination, and SUMOylation of lysine residues – Phosphorylation of serine residues – Methylation of arginines – Most frequent histone lysine modifications [71]: methylation of histone H3 at Lys9 (H3-K9) or Lys27 (H3-K27) is associated with gene silencing – Methylation or acetylation of histone H3 at Lys4 (H3–K4) or acetylation of H3 at Lys27 (H3-K27) is associated transcriptional activation | |
Enzymes involved in these processes: – Histone acetyltransferases (HATs) – Histone deacetylases (HDACs) – Histone methyltransferases – Methyl-binding domain protein MECP2 | • HATs can be divided into several families, including the PCAF/Gcn5, p300/CBP, MYST, SRC, TAFII250, HAT1, and ATF-2 families • HDACs are classified into four groups (I–IV) |
The transcriptional coactivator, peroxisome proliferative activated receptor gamma coactivator 1 alpha (PPARGC1A) , coordinates the regulation of genes involved in energy metabolism by controlling transcriptional programs of mitochondrial biogenesis, adaptive thermogenesis, and fatty acid beta-oxidation [64]. In fact, its tissue specificity pattern of expression is located mainly in the heart, skeletal muscle, liver, and kidney. Interestingly, the protein encoded by this gene is involved in controlling blood pressure, regulating cellular cholesterol homoeostasis, and in the development of obesity, and altered signaling of PPARGC1A contributes to glucose intolerance, insulin resistance, and type 2 diabetes [64, 65]. We focused on methylation of 5-methylcytosine in dinucleotide CpG, which is associated generally with gene silencing, and measured the level of DNA methylation of three putative methylation target sites in the promoter of the PPARGC1A (located at positions -513, -519, and -615 relative to transcriptional start site) [66]. Interestingly, we demonstrated that the methylation status of the PPARGC1A promoter in the liver of patients with NAFLD is significantly associated with plasma fasting insulin levels and homeostasis model assessment of IR (HOMA-IR), regardless of the liver disease severity [66]. As expected, we observed that the methylation status of the PPARGC1A promoter was significantly associated with fatty liver as a disease trait, showing that a higher proportion of the alleles was methylated in NAFLD patients compared with in the liver of control subjects [66]. We also observed that liver PPARGC1A mRNA abundance was inversely correlated with the methylation levels of PPARGC1A promoter CpGs, suggesting that the methylation of at least the three explored sites in the promoter repressed the transcriptional gene activity efficiently [66].
An interesting study explored the pre- and post-bariatric changes in the methylation profile of nine genes coding for enzymes that regulate intermediate metabolism and insulin signaling in the liver of morbidly obese patients with NAFLD [67]. The most remarkable finding of this study is that therapeutic intervention partially reverted NAFLD-associated methylation changes; for instance, the gene encoding protein-tyrosine phosphatase epsilon (PTPRE) showed both differential expression and differential methylation before and after bariatric surgery [67]. Moreover, the authors observed that the insulin-like growth factor-binding protein 2 (IGFBP2) locus was hypermethylated and its mRNA downregulated in NASH.
Murphy and colleagues, who recently did global methylation profiling of liver samples of NAFLD patients at different stages of disease severity by using the Illumina HumanMethylation450 BeadChip platform, observed that patients with advanced NAFLD had a signature of differentially methylated CpG sites that allow discrimination between advanced versus mild disease [68]. Indeed, the authors showed that advanced NAFLD has a relative hypomethylation state (11 % of 52,830 CpG sites) compared with mild NAFLD, specifically in genes associated with tissue repair, for instance, FGFR2 (a fibroblast growth factor receptor family member), genes of the collagen (COL1A1, COL1A2, COL4A1, and COL4A2) and laminin families, and many chemokines [68]. Of note, genes involved in pathways that generate methyl groups, including methylenetetrahydrofolate dehydrogenase 2 (MTHFD2), were significantly hypomethylated in advanced NAFLD [68].
Finally, we recently described a novel disease mechanism associated with NAFLD progression that involves epigenetic changes of mitochondrial DNA (mtDNA) [69]. In our study, we explored the status of cytosine methylation of liver mtDNA in target regions of the mtDNA genome for the first time. We observed that the methylation levels of mitochondrial NADH dehydrogenase 6 (MT-ND6), the gene that encodes for a key enzyme of complex 1 of the oxidative-phosphorylation (OXPHOS) chain, were higher in the livers of NASH patients and that there was a clear decrease in the protein levels and changes in mitochondrial morphology, suggesting that the methylation status of this mitochondrial gene plays a role in the phenotypic switching from SS to NASH [69]. To contrast with the hypothesis that epigenetic modifications might be reversible by intervention, we explored whether the observed changes were associated with interventional programs, observing that physical activity modulates the methylation status of MT-ND6 [69].
On the other hand, it is known that chronic ethanol intake lowers the hepatocellular S-adenosylmethionine-to-S-adenosylhomocysteine ratio and significantly impairs liver methylation reactions [70]. Recently, Kharbanda and colleagues showed that chronic alcohol consumption is associated with a decrease in the hepatic methylation capacity, causing liver injury. Of note, the authors suggested that dietary intervention might be recommended to subjects with AFLD, as exposure to methyl-group consumers, such as guanidinoacetate, can aggravate the liver toxicity of ethanol [70].
Furthermore, it was shown that ethanol increases acetylation of H3-Lys9 through modulation of HAT(s) [71]. A comprehensive review on ethanol and epigenetic modifications was published recently [72].
In summary, epigenetic modifications emerged as an interesting target of therapeutic intervention in chronic and prevalent human diseases as they offer a unique framework of reversible mechanisms that modulate cellular transcriptional machinery.
The Role of Mitochondrial Dysfunction in the Development and Disease Progression of NAFLD and AFLD
Resembling our observations about the critical role of PPARGC1A on the development of NAFLD, in experimental models, Lieber and coworkers demonstrated that alcohol reduces the hepatic expression of PPARGC1A mRNA by about 50 % [73], reinforcing the importance of master regulators of metabolism in the energy sensing system of the liver that also lead to mitochondrial dysfunction and damage [66, 69, 73]. In agreement with these observations, Han et al. reported that liver mitochondria undergo dynamic alterations following chronic alcohol feeding in mice that include increased mitochondrial NAD+ and NADH levels with enhanced mitochondrial biogenesis in the liver to adapt to metabolic stress [74]. Our group observed a similar finding in rats, demonstrating that in rodents, metabolic insults, like high-fat diet, promote an increase in liver mitochondrial biogenesis in response to hypoxia via HIF-1alpha, probably to enhance the mitochondrial function and to accommodate the metabolic stress [75]. Nevertheless, once the NAFLD is established in humans and the disease progresses to NASH, mitochondrial DNA content is significantly reduced in the liver, suggesting that the advanced disease is associated with a loss of the adaptive mechanism for the metabolic demands [66].
Related posts:
 Nonalcoholic Fatty Liver Disease in Children
Diagnostic Considerations and Clinical End Points for Nonalcoholic Steatohepatitis
Hepatic and Extrahepatic Malignancies in NAFLD
Nonalcoholic Fatty Liver Disease in Children
Diagnostic Considerations and Clinical End Points for Nonalcoholic Steatohepatitis
Hepatic and Extrahepatic Malignancies in NAFLD
 Clinical Features, Disease Modifiers, and Natural History of Alcoholic Liver Disease
Clinical Features, Disease Modifiers, and Natural History of Alcoholic Liver Disease
 Animal Models of Alcoholic Liver Disease
Animal Models of Alcoholic Liver Disease
 Epidemiology and Risk Factors for Alcoholic Liver Disease
Epidemiology and Risk Factors for Alcoholic Liver Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


