CHAPTER 31 Gastrointestinal Carcinoid Tumors (Gastrointestinal Neuroendocrine Tumors) and the Carcinoid Syndrome
In 1888, Lubarsch described what is now recognized to be the entity of carcinoid tumors when he reported the autopsy finding of two patients with multiple tumors in the distal ileum.1 The term carcinoid was introduced by Oberndorfer in 1907 in his description of a class of malignant tumors that behaved less aggressively than the more common adenocarcinomas of the gastrointestinal (GI) tract.2 The traditional classification of carcinoids, based on their embryonic origin into foregut, midgut, and hindgut carcinoids, has been gradually abandoned.3 A tumor biology–based classification system introduced by the World Health Organization (WHO) in 2000 has greater applicability, although more recent amplification and revision of this system by the European Neuroendocrine Tumor Society (TNM classification) appears likely to become the standard (Table 31-1).4,5
Table 31-1 TNM Classification for Endocrine Tumors*
| A. Gastric Tumors | |
| TX | Primary tumor cannot be assessed |
| T0 | No evidence of primary tumor |
| Tis | In situ tumor, dysplasia (<0.5 mm) |
| T1 | Tumor invades lamina propria or submucosa and is ≤1 cm |
| T2 | Tumor invades muscularis propria or subserosa or is >1 cm |
| T3 | Tumor penetrates serosa |
| T4 | Tumor invades adjacent structures |
| NX | Regional lymph nodes cannot be assessed |
| N0 | No regional lymph node metastases |
| N1 | Regional lymph node metastases |
| MX | Distant metastases cannot be assessed |
| M0 | No distant metastases |
| M1 | Distant metastases |
| B. Tumors of the Duodenum, Ampulla, and Proximal Jejunum | |
| TX | Primary tumor cannot be assessed |
| T0 | No evidence of primary tumor |
| T1 | Tumor invades lamina propria or submucosa and is ≤1 cm |
| T2 | Tumor invades muscularis propria or is >1 cm |
| T3 | Tumor invades pancreas or retroperitoneum |
| T4 | Tumor invades peritoneum or other organs |
| NX | Same as for gastric |
| N0 | Same as for gastric |
| N1 | Same as for gastric |
| MX | Same as for gastric |
| M0 | Same as for gastric |
| M1 | Same as for gastric |
| C. Tumors of Distal Jejunum and Ileum | |
| TX | Primary tumor cannot be assessed |
| T0 | No evidence of primary tumor |
| T1 | Tumor invades mucosa or submucosa and is ≤1 cm |
| T2 | Tumor invades muscularis propria or is >1 cm |
| T3 | Tumor invades subserosa |
| T4 | Tumor invades peritoneum/other organs |
| NX | Same as for gastric |
| N0 | Same as for gastric |
| N1 | Same as for gastric |
| MX | Same as for gastric |
| M0 | Same as for gastric |
| M1 | Same as for gastric |
T, primary tumor (for any T, an “m” is added for multiple tumors; N, regional lymph nodes; M, metastases).
* European Neuroendocrine Tumor Society.
Adapted from Solcia E, Kloppel G, Sobin L. Histological typing of endocrine tumours. In: Verlag S, editor. World Health Organization Histological Classification of Tumours. 2nd ed. New York: Springer; 2000. p 38; and Rindi G, Kloppel G, Alhman H, et al. TNM staging of foregut (neuro)endocrine tumors: A consensus proposal including a grading system. Virchows Arch 2006; 449:395-401.
Carcinoids arise from cells of the diffuse neuroendocrine system and can arise almost anywhere within the gastrointestinal tract.6 Carcinoids, also called gastrointestinal neuroendocrine tumors (GI NETs), are related to medullary carcinoma of the thyroid, pheochromocytoma and pancreatic neuroendocrine tumors. GI NETs synthesize bioactive amines and peptides, including neuron-specific enolase (NSE), 5-hydroxytryptamine (5-HT, or serotonin), and 5-hydroxytryptophan (5-HTP). They also secrete peptides such as chromogranin A, pancreatic polypeptide, calcitonin, tachykinins (neurokinin A and substance P), and various growth factors, such as transforming growth factor-β (TGF-β), platelet-derived growth factor (PDGF), endocrine growth factor (EGF), fibroblast growth factor (FGF), and the vascular endothelial growth factor (VEGF) family of growth factors, including their receptors.7–9 Of clinical relevance is the observation that several different genes and genetic divergences related to tumor development are evident. GI NETs can be sporadic (nonfamilial) or part of a familial syndrome, such as von Hippel-Lindau syndrome or neurofibromatosis (NF). GI NETs comprise 0.5% of all malignancies and, as shown in Figure 31-1, their incidence has increased substantially over the last several decades.10,11 In this chapter, we will use the more familiar term carcinoid, although the term GI NET is the most appropriate, but not as well established.
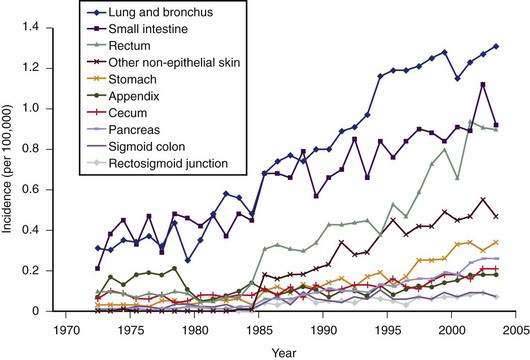
Figure 31-1. Incidence of different subtypes of neuroendocrine tumors, 1970 to 2005, from the Surveillance Epidemiology and End Results (SEER) data base.10,11 Note the significant increase in most subtypes of neuroendocrine tumors since the 1970s.
(From Modlin IM, Oberg K, Chung DC, et al. Gastroenteropancreatic neuroendocrine tumours. Lancet Oncol 2008; 9:61-72.)
Carcinoid tumors occur most frequently in the GI tract (67%), with the bronchopulmonary system being the second most common location (25%), followed by considerably less frequent locations, such as the ovaries, testes, and hepatobiliary system.12 The most common location in the GI tract is the ileum (17%). The overall incidence of carcinoid tumors is difficult to determine, because it appears likely that most tumors remain asymptomatic. An autopsy study has estimated the annual incidence to be 8.4/100,000 people.13 The SEER database from 1973 to 2004 indicated an annual incidence of 2.0 to 2.5/100,000/year, with a 3.5% annual increase over this time period.10 Ileal tumors have specifically increased in prevalence—in white males (by 274%), black males (500%), white females (213%), and black females (286%), respectively. A recent evaluation of the SEER database comprising 35,825 cases in the United States has indicated an annual incidence of 5.25/100,000/year for all types of neuroendocrine tumors and a prevalence of 35/100,000.14 The annual incidence of gastrointestinal carcinoids was 2.53/100,000. Overall, the incidence of gastrointestinal carcinoids is higher in African Americans (4.5/100,000) compared with white Americans (2.5/100,000) with striking differences in certain sites, particularly the rectum. As a result of the increasing incidence and prevalence, gastrointestinal neuroendocrine tumors represent a substantial clinical problem. The increasing incidence is probably mostly to the result of the introduction of better diagnostic tools (imaging and immunohistochemical), as well as greater awareness by pathologists and clinicians. There are no known environmental risk factors for carcinoids.
CLINICAL FEATURES
An early and accurate diagnosis is often delayed by four or five years because most small intestinal carcinoids (Fig. 31-2) are small, initially asymptomatic, or misdiagnosed as conditions such as allergy or irritable bowel syndrome. Progressive growth of the tumor may cause vague abdominal discomfort because of intermittent intestinal obstruction. As the tumor gets larger, invading the intestinal wall and occluding the gut lumen, emergency clinical presentations of an acute abdomen (intussusception, obstruction, perforation, bleeding) may arise because of local tumor mass effect or tumor-induced fibrosis.15–18 The frequency of intestinal obstruction that is secondary to a gastrointestinal NET-associated mesenteric fibrosis ranges from 42% to 66%.18,19 Another characteristic feature of these patients is vascular elastosis (thickening of the vessel wall), resulting in ischemic changes of the gut. The cause of this condition is unclear, but a local effect of serotonin or other tumor products, such as TGF-α, have been postulated to have a direct trophic effect on smooth muscle and fibroblasts in the vessel wall. Both mesenteric fibrosis and elastosis may cause abdominal angina.15 A rare cutaneous manifestation of ileal carcinoids is a fibrotic, scleroderma-like condition mostly affecting the lower extremities.20 As the disease advances, gastrointestinal carcinoids frequently metastasize locally to mesenteric lymph nodes and to the liver. A classic carcinoid syndrome is relatively uncommon (10% to 15%), typically consisting of diarrhea, cutaneous flushing, bronchoconstriction and right-sided heart failure.21,22 As many as 15% to 25% of gastrointestinal carcinoids exhibit a synchronous or metachronous association with other tumors, usually adenocarcinomas of the colon.23–25 This may reflect the activity of growth factors produced by the carcinoid tumor. A relatively large percentage of GI NETs are multicentric; for example, up to 33% of carcinoids in the small intestine are multicentric.24–26
PATHOLOGY
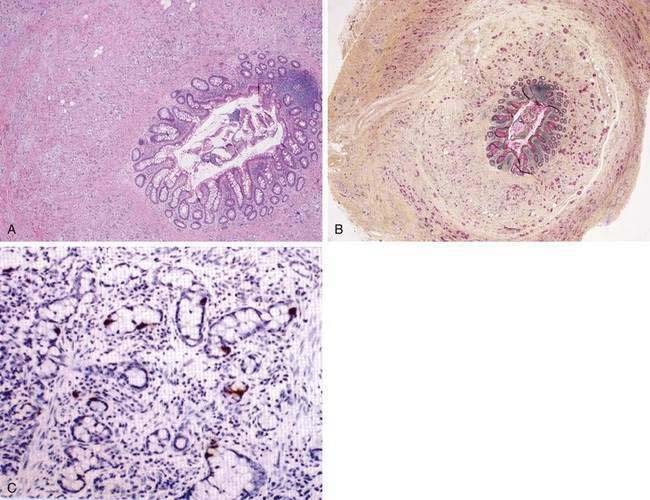
The most common histopathologic type of intestinal carcinoid is the enterochromaffin (EC) cell carcinoid, accounting for more than 98% of cases. The EC cell carcinoid is defined by its argentaffin staining properties, serotonin production, and typical pleomorphic secretory granules. Morphologically, EC cell carcinoids are characterized by medium-sized tumor cells arranged in an organoid pattern and showing only mild to moderate atypia. Tumor necrosis is absent and the mitotic rate is low (<2 mitoses/10 high-powered field [HPF]). Distinct growth patterns have been described in gastrointestinal carcinoids: (1) nodular or insular pattern; (2) trabecular pattern; (3) acinar and tubular pattern; and (4) atypical solid pattern. Mixed patterns may be seen. EC cell carcinoids of the jejunum and ileum predominantly display an insular growth pattern (93%; Fig. 31-3) and, less frequently, mixed insular and glandular (5%) or trabecular (2%) growth patterns. EC cell carcinoids invade the submucosa, muscularis propria, or mesentery. Invasion of lymphatics and veins, or perineural growth, can be seen. EC cell carcinoids may produce multiple hormones. Most of the tumors (>90%) secrete serotonin and tachykinins (substance P, neurokinin A). Production of other hormones such as gastrin, glucagon, cholecystokinin, calcitonin, somatostatin, or adrenocorticotropic hormone (ACTH) can be demonstrated in less than 5% of cases. Neuroendocrine markers (proteins associated with large dense core vesicles or synaptic-like microvesicles) are abundantly expressed in EC cell carcinoids and can be used to confirm the neuroendocrine phenotype of the these tumors.24,25,27 Chromogranin A, synaptophysin, synaptic vesicle protein 2, neuron-specific enolase, and Leu7 are expressed by 92% to 100% of jejunoileal carcinoid tumors. Expression of the vascular monoamine transporters 1 and 2 (VMAT-1 and -2) has been demonstrated in more than 90% of tumors and is related to the production and storage of serotonin in EC cell carcinoids. Other markers identified in EC cell carcinoids include cytokeratins 8 and 18, carcinoembryonic antigen (CEA), and prostatic acid phosphatase. High expression of the intestinal transcription factor CDX2 has been demonstrated in EC cell carcinoids and may become a useful marker of intestinal origin.6,27–33
Carcinoids (GI NETs) arising in the duodenum and jejunum include gastrin, somatostatin, and EC cell tumors and gangliocytic paraganglioma. Gastrin cell tumors (gastrinomas) are the most common in the duodenum, might be nonfunctioning or functioning (Zollinger-Ellison syndrome [ZES]), and associated with the multiple endocrine neoplasia type I (MEN-I) syndrome (see Chapter 32). Tumors are mainly located in the first and second parts of the duodenum. A small proportion of tumor cells may produce other hormones in addition to gastrin, such as cholecystokinin (CCK), somatostatin, pancreatic polypeptide, neurotensin, and insulin. Morphologically, tumor cells are uniform, with rounded nuclei and abundant cytoplasm. The growth pattern is usually trabecular or cribriform. Necroses are absent and the mitotic rate is low. Most of the tumor cells are positive for neuroendocrine markers, such as chromogranin A, synaptophysin, Leu7, and NSE.34–36
Somatostatin cell tumors (somatostatinomas) represent the second most frequent histopathologic type in the duodenum, accounting for 15% to 27 % of duodenal GI NETs (see Chapter 32). Somatostatin cell tumors are preferentially localized to the periampullary region. They are identified by their content of somatostatin and typical large electron-dense secretory granules. A subset of tumor cells may also contain calcitonin, pancreatic polypeptide, and ACTH. Morphologically, tumors are characterized as a mixture of tubular-glandular, insular, and trabecular growth patterns and the presence of psammoma bodies. Tumor cells are uniform and the mitotic rate is low.34,35,37,38
Gangliocytic paraganglioma represent the third most frequent histopathologic type in the duodenum and account for 6% to 9% of duodenal carcinoids. The tumors are preferentially located in the periampullary region and are not associated with familial syndromes. Gangliocytic paragangliomas are identified by their characteristic morphology, which includes a mixture of three different cell types—spindle cells, epithelial cells, and ganglion cells. Spindle cells are arranged in fascicles and stain positive for S-100. Epithelial cells represent the endocrine component and are positive for pancreatic polypeptide, somatostatin, and chromogranin A. Ganglion cells represent the neural component and are positive for synaptophysin and NSE. Gangliocytic paraganglioma are regarded as benign hamartomatous tumors, derived from the ventral pancreatic primordium.39–42
Endocrine tumors of the stomach are composed of cells with features similar to those of the normal endocrine cell counterparts. The most representative cell types are the histamine-producing enterochromaffin-like (ECL) cell in the corpus and fundus and the gastrin-producing G cell in the antrum, representing about 50% of all endocrine cells at both sites. The other endocrine cell types present throughout the stomach are somatostatin-producing (D), ghrelin-producing (P), and serotonin-producing enterochromaffin (EC) cells. All cell types may be found in gastric tumors, but preneoplastic lesions are known to be comprised mostly of ECL cells. Antral G cell hyperplasia is often observed in chronic atrophic gastritis (see Chapter 51), but it is not considered a preneoplastic lesion. More than 90% of gastric endocrine tumors are well-differentiated tumors–carcinomas (WDET/C), according to the WHO classification. More than 90% of the tumors occur in the corpus-fundus area and are composed of ECL cells, with only rare EC, G, or P cell tumors. Poorly differentiated neuroendocrine carcinomas of the GI tract are highly malignant tumors.43–50 In general, the cytosol markers NSE and PGP 9.5, and the small synaptic-like vesicle marker synaptophysin, are expressed in the poorly differentiated tumors in contrast to well-differentiated neuroendocrine tumors. Chromogranin A and tissue-specific hormones are generally absent or sparse in poorly differentiated neuroendocrine carcinomas. Hormones and chromogranin A are stored in large dense core vesicles, which are rarely observed in poorly differentiated gastric and intestinal tumors.
MOLECULAR GENETICS
Studies have shown that development of various types of GI NETs might involve different genes associated with distinct abnormalities, including point mutations, deletions, methylations, and chromosomal losses and gains (see Chapter 3). The menin gene, a tumor suppressor gene, encodes a protein of 610 amino acids; mutations of this protein cause most cases of MEN-I and a smaller proportion of sporadic and gastric duodenal endocrine tumors.51 Menin is mainly a nuclear protein, but in dividing cells, it interacts in the cytoplasm with several proteins that control transcription, regulation of genome stability, and cell division. Small intestinal carcinoid tumors show deletions on chromosome 18.52,53 Recent studies have also demonstrated overexpression of the neoplasia-related genes NAP1L1 (mitotic regulation), MAGE D2, and MTA1. These genes are thought to regulate the malignant potential of these tumors and their propensity to metastasize.54 Hindgut neuroendocrine tumors express receptors for TGF-α and EGF.51
CLASSIFICATION AND SUBTYPES
Carcinoid tumors were previously classified according to their embryologic region of origin into foregut, midgut, or hindgut tumors. This classification has now largely been abandoned and a WHO classification system is generally accepted. It has been recently updated by the European Neuroendocrine Tumor Society to a TNM and grading system (see Table 31-1).4,5
ESOPHAGUS
Carcinoid tumors of the esophagus are rare.55 However, in a series from Japan, esophageal carcinoids constituted 27% of all GI carcinoids, more frequent than ileal and appendiceal carcinoids.42 The male-to-female ratio was 3.3, indicating a male preponderance for this type of carcinoid tumor. Dysphagia is the most common presenting symptom and is usually localized to the distal portion of the esophagus. Esophageal carcinoids may occur in conjunction with adenocarcinomas arising from Barrett’s esophagus.56
STOMACH
A trend for an increased incidence of gastric neuroendocrine tumors ha been noted in surgical and endoscopic series. In the Surveillance, Epidemiology, and End Results (SEER) database, there was an increase in gastric neuroendocrine tumors, from 2.4% to 8.7% of all GI carcinoids, between 1950 and 1999.12 In mainly endoscopic series, much higher relative incidences were reported, ranging from 11% to 41% of all gastrointestinal neuroendocrine tumors.57 However, the increased relative incidence of gastric neuroendocrine tumors must be put into the context of a wider incremental trend for all types of gastrointestinal well-differentiated neuroendocrine tumors (carcinoids). Gastric neuroendocrine tumors are currently classified as well-differentiated and poorly differentiated lesions on the basis of the differentiation status of the tumor cells. The most common types are tumors derived from histamine-producing ECL cells in the corpus-fundus region, followed by gastrin producing G cell tumors of the antrum.57–59 Rare types of gastric carcinoid are those producing gastrin, ghrelin, and serotonin.43,44
Three clinicopathologic subtypes of ECL cell tumors are recognized (Table 31-2). The type I ECL tumor is associated with diffuse corpus-restricted chronic atrophic gastritis (see Chapter 51). Type II is associated with MEN-I, ZES, and hypertrophic gastropathy. Type III tumors are sporadic are not associated with any distinctive gastric pathology.43,58,59 Types I and II tumors share in common hypergastrinemia, whereas type III tumors are independent of any overt hormonal imbalance. Type I ECL tumors account for the largest fraction of well-differentiated neuroendocrine tumors of the stomach, are especially prevalent in older women, and are associated with antral G cell hyperplasia. Frequently, multiple and multicentric lesions are present and are generally small and limited to the mucosa or submucosa. Metastases are rare and survival is excellent.45 Type II ECL tumors are rare and account for only 6% of gastric carcinoids. Type II tumors arise in adult patients of both genders who have hypergastrinemia, hypertrophic hypersecretory gastropathy, and ECL cell hyperplasia. The ECL tumors are often multiple, multicentric, small in size, and limited to mucosa and submucosa. Despite metastases to local lymph nodes, the survival is excellent and tumor-related death is rare.57 Type III ECL cell tumors are usually single, isolated growths arising in the stomach, without any significant underlying gastric pathology. In one series, they accounted for 14% of all gastric carcinoids. They are more common in men, usually in their sixth decade, and without hypergastrinemia and gastrin-dependent ECL cell hyperplasia. The tumor size may be significantly larger than in types I and II (mean, 3.2 cm), with invasion of the stomach wall and metastases in more than 50% of patients.57 Predictors of malignancy in well-differentiated gastric carcinoids include size, histologic grading, mitotic count and Ki67 index, and p53 overexpression. Poorly differentiated endocrine tumors of the stomach are aggressive, usually large carcinomas and develop with no site predilection in the stomach. They usually occur in patients in the sixth to seventh decade of life. These poorly differentiated tumors are often metastatic at time of diagnosis; their prognosis is invariably poor.

PANCREAS
The differentiation between pancreatic carcinoid and other pancreatic neuroendocrine tumors is primarily a matter of definition (see Chapter 32 for a more detailed discussion). Maurer and colleagues have defined a pancreatic carcinoid as a tumor with typical histologic features of an NET along with evidence of increased serotonin metabolism.60 Using this definition, they found only 29 cases in the literature between 1966 and 1996. In a more recent publication from Japan, Soga found 156 cases of pancreatic carcinoids among 11,343 cases of neuroendocrine tumors (1.4%).61 These tumors were characterized by a high metastatic rate (66%), a large tumor size (averaging almost 7 cm), and a relatively high incidence of the carcinoid syndrome (23%). Serotonin was detected by immunohistochemical methods in 93% of cases. The five-year survival rate was extremely low (29%) when compared with small intestinal carcinoid (82%). Pancreatic carcinoids, like other pancreatic tumors, tend to present later when compared with carcinoids in other locations. Abdominal pain, diarrhea, and weight loss are the most common presenting symptoms.
DUODENUM AND AMPULLA OF VATER
About 4% of all gastrointestinal carcinoids occur in the duodenum and duodenal carcinoids account for 11% of all intestinal carcinoids. The relatively frequency of duodenal carcinoids increased from 3.6% to 16% of all intestinal carcinoids from 1973 to 2002. The annual incidence of duodenal carcinoid is 0.07/100,000. It is higher in males than in females and is higher in blacks than in whites.10–12 Duodenal carcinoids present at a mean age of 48 to 62 years. Most duodenal carcinoids give rise to symptoms related to local growth, such as obstruction, jaundice, abdominal pain with or without pancreatitis, gastrointestinal bleeding, nausea, and vomiting.35,62 A minority of patients with duodenal carcinoids (<10%) present with symptoms and/or signs of hormone overproduction. ZES occurs in approximately 10% of patients with duodenal carcinoids (see Chapter 32). The carcinoid syndrome may occur (4%) and, in rare cases, Cushing’s syndrome and acromegaly may be seen.
Gastrin (G) cell tumors represent the most frequent histopathologic type, accounting for 50% to 60% of all duodenal carcinoids (Table 31-3). G cell tumors can be nonfunctioning or functioning (Zollinger-Ellison syndrome) and may be associated with the MEN-I syndrome.36,62 Somatostatin cell tumors represent the second most frequent histopathologic type (15% to 27%).38 Somatostatin cell tumors are preferentially localized to the periampullary region. They are identified by their content of somatostatin, but a subset of tumor cells may also contain calcitonin, pancreatic polypeptide, and ACTH.
Table 31-3 Types of Duodenal and Ampullary Carcinoid Tumors
| CELL OF ORIGIN/TUMOR TYPE | RELATIVE FREQUENCY (%) |
|---|---|
| Gastrin (G) cell* | 50-60 |
| Somatostatin (D) cell | 15-77 |
| Gangliocytic paraganglioma | 6-9 |
| EC, others | <5 |
EC, enterochromaffin.
* Functional (Zollinger-Ellison syndrome) or nonfunctional.
Gangliocytic paragangliomas represent the third most frequent histopathologic type and account for 6% to 9% of duodenal carcinoids.39,40,62 The tumors are preferentially located in the periampullary region and are not associated with familial syndrome. The tumors are regarded as benign hamartomatous tumors, derived from the ventral pancreatic primordium. EC cell tumors of the duodenum are rare, and have the same histopathologic features as ECL cell tumors in the lower jejunum and ileum.
The overall five-year survival rate for duodenal carcinoids is reported to be 84%.10–12 However, survival rates vary with histopathologic type, extent of disease, presence of hormonal syndrome, and genetic background. In patients with Zollinger-Ellison syndrome, duodenal localization of the primary tumor carries a much better prognosis than pancreatic localization (see Chapter 32), with 10-year survival rates of 94% and 55%, respectively.10–12,62
SMALL INTESTINE
Forty-four percent of all gastrointestinal carcinoids arise in the small intestine. The ileum is the most frequent location for gastrointestinal carcinoids.10–12,14 The annual incidence of intestinal carcinoids is 0.63/100,000 and 0.04 and 0.31/100,000 for jejunal and ileal carcinoids, respectively. The incidence of intestinal carcinoids has increased four- to fivefold during the past 30 years. Small intestinal carcinoids result from malignant transformation of EC cells. A considerable percentage (17% to 29%) of the patients develop noncarcinoid GI and non-GI tumors, mainly adenocarcinomas of the gastrointestinal tract, suggesting a common pathogenic mechanism for carcinoid and noncarcinoid tumors.23 Familial occurrence of intestinal carcinoid is rare, although a few families have been reported. Jejunal and ileal carcinoids are also rarely associated with MEN-I.27
Patients with carcinoid tumors of the jejunum and ileum have a mean age of 55 to 63 years at the time of diagnosis.15,21 Usually, there is a delay of four to five years for diagnosis of this tumor.17 The most frequent presentations are abdominal pain (16% to 41%) and episodic small bowel obstruction (24% to 41%), which are usually of long duration. Diarrhea, gastrointestinal bleeding, flushing, and weight loss are presenting symptoms in 4% to 30%. The carcinoid syndrome is encountered in 5% to 18% of patients.16,63
Carcinoid tumors occur all along the jejunum and ileum, but increase in frequency distally, with their highest frequency in the terminal ileum.12,13 There is a single primary tumor in 74% of patients, whereas multiple tumors occur in 26% of patients.26 Clonality studies have indicated that multiple tumors are generated by the metastasis of a single primary tumor to different locations in the intestine.64 The size of the primary tumor ranges from 0.3 to 5.5 cm (mean, 2.5 cm). Intestinal carcinoids are usually associated with pronounced fibrosis and tissue scarring, which may cause intestinal obstruction and infarction. The desmoplastic stromal reaction surrounding intestinal carcinoids has been attributed to secretion of hormones and growth factors by tumor cells. Small intestinal carcinoids may produce multiple hormones. Most secrete serotonin and tachykinins (substance P and neurokinin A). Production of other hormones, such as gastrin, glucagon, cholecystokinin, calcitonin, somatostatin, and ACTH, can be demonstrated in less than 5% of cases.7,63
Data from the SEER registry for period 1973 to 2002, summarized in Table 31-4, reveal an overall five-year survival rate for intestinal carcinoids of 63% and a five-year survival rate of 84%, 72%, and 43% for localized, regional, and distant (metastatic) disease, respectively.10,11,16 However, five-year survival rates of 69% have been reported for patients with intestinal carcinoids metastatic to the liver who were given active interventional treatment.65 Risk factors for mortality include distant metastases, carcinoid syndrome, and female gender.66 Histopathologic markers shown to be associated with poor prognosis are a solid growth pattern and a Ki67 index above 1%.67

APPENDIX
The appendix is one of the most common sites in the GI tract for the development of a carcinoid tumor. The frequency of carcinoid tumors in nonselected series of appendectomy specimens ranges from 0.2% to 0.9%, but more recent data have suggested that the overall trend is toward a decreased prevalence.68,69 It is reported that appendiceal carcinoids occurs twice as often in women as in men. This observation may partly reflect the frequency of incidental appendectomy during gynecologic and gallbladder surgery but the female-to-male ratio is 2 : 1, even after correction for these factors.
Carcinoid tumors of the appendix have been classified based on their histogenesis, cell types, and histologic pattern, as well as their clinical behavior. All typical appendiceal carcinoid tumors are clinically silent and are discovered incidentally during surgery performed for symptoms of acute appendicitis or during incidental appendectomy. Most typical appendiceal carcinoids occur in the tip of the organ. They are usually small (<1 cm) and rarely exceed 2 cm in diameter. Metastases are rare in typical appendiceal carcinoid.68,69 The overall prognosis for EC cell appendiceal tumors is also favorable, with a 10-year disease specific survival of more than 98%. The incidence of metastases is less than 1% if tumor size is between 1 and 2 cm. Thus, appendectomy alone is likely to be adequate treatment for the most typical appendiceal carcinoid tumors that are smaller than 2 cm.70,71 Conversely, right hemicolectomy should be considered in selected patients with a carcinoid more than 2 cm in diameter, with a tumor that extends into the muscularis propria, and those with a positive resection margin, associated perforation, or evident lymph node metastases. Typical appendiceal carcinoids have the best prognosis of all types of carcinoids, which probably reflects the anatomic site and biologic behavior of the tumor itself; early detection and removal are beneficial.
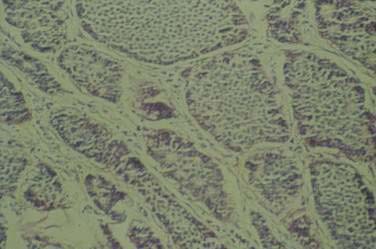
In contrast to typical carcinoid tumors of the appendix, goblet cell carcinoids of the appendix (Fig. 31-4) have a mixed phenotype, with partial neuroendocrine differentiation and intestinal-type goblet cell morphology.72 The mean age at diagnosis is 50 years, with a range of 29 to 80 years. Goblet cell carcinoids constitute a family of appendiceal carcinoids with various morphologic features and malignant behavior. Some of them are histologically aggressive neoplasms, such as adenocarcinomas of the GI tract. The tumors demonstrate neuroendocrine differentiation, with positive staining for chromogranin and synaptophysin, although the staining is often focal and always much less extensive than in classical carcinoid tumors. The most common and distinctive feature is mucin-containing goblet-shaped epithelial cells arranged in round or oval clusters.73,74 The overall five-year survival rate for different subtypes of goblet cell carcinoids is 42%, with a three-year survival rate of only 17% for the most malignant subtypes.