Gastrointestinal Cancer: Cancer Genetics
Steven M. Lipkin
Kenneth Offit
Introduction
Clinical genetic services for gastrointestinal (GI) cancer help the medical community effectively identify families with genetic syndromes. When properly diagnosed, these patients and their relatives can benefit from early detection through frequent cancer surveillance, risk modification, chemoprevention, and participation in screening and treatment clinical trials. The goal is to detect tumors early and improve survival and quality of life of families carrying these genetic mutations. At the same time, a related goal is to spare their relatives who are not at increased cancer risk the discomfort and expense of frequent cancer screening surveillance, or even unnecessary surgical resection of the relevant organs. By providing more precise and personalized prediction of cancer risk, clinical genetics can prevent cancer in patients and their families affected with genetic syndromes and more effectively can manage medical resources.
Germline mutations in genes that predispose to colorectal, small bowel, biliary tract, and pancreatic adenocarcinomas have been identified. Genetics has had a significant impact in identifying individuals who can be targeted for intensive screening surveillance and, when indicated, surgery. The contribution of germline mutations in known susceptibility genes, as described in this chapter, is generally believed to be 25% or less for GI malignancies. However, with each year more atypical presentations of the known syndromes appear that extends their spectrum. The contribution of hypomorphic (i.e., partial loss of function) mutations causing cancers at lower rates of penetrance, perhaps clinically indistinguishable from sporadic forms, is likely to be more common than currently appreciated, and the total contribution of GI cancer attributable to germline mutations revised upward.
Because many of these GI cancer genetic risk–related deaths are preventable with early detection, it is a critical ethical (and increasingly legal) imperative for primary care physicians to be familiar with and recognize cancer-related genetic syndromes. Eliciting the existence of early onset cancers in the personal or family medical history, constellations of cancers in a family that fit known syndromes, or tumor pathology associated with known syndromes, as described here, can be a lifesaving intervention. Similarly, a basic familiarity for the primary care provider during the physical exam of the physical stigmata associated with some of these syndromes (e.g., pigmented mucocutaneous lesions or extranumerary teeth) can help identify the individuals at risk. If the primary care physician were uncertain about the significance of patient history and physical exam findings, or associated pathological findings such as rare tumor subtypes (e.g., hamartomatous polyps), it would be important to refer the patient to qualified genetic professionals. These professionals include certified genetic counselors, medical geneticists, oncologists, and gastroenterologists, among others. If not available readily in the community practice setting, it is the responsibility of the primary care provider to refer to providers of these services. With reference to the interpretation of genetic tests, and indeed how to distinguish genetic variants from bona fide deleterious mutations, if the physician ordering the test were to be in doubt, consultation with genetic professionals should be performed. In terms of patient surveillance and management post identification of a genetic mutation related to tumors of the GI tract and related sites, these patients can be managed by the GI specialist, qualified genetic professionals, or a thoughtful and motivated primary care physician. Useful resources about specific genetic tests and the clinical presentations of genetic syndromes can be found on the Genetests and Online Mendelian Inheritance in Man Web sites.
Diagnosis of a genetic syndrome is made by mutation analysis of germline genomic DNA for specific candidate genes. In the United States, these tests currently cost about $200 to $3,000, depending on the analysis performed. A 5- to 10-cc blood sample provides ample DNA for testing. On occasion, other tissue sources, such as skin fibroblast biopsies, are used. Typically, all the exons in the coding region of the gene(s) to be analyzed are polymerase chain reaction (PCR) amplified and sequenced. If a gene variant were to truncate the candidate protein prematurely, such as by the creation of a new stop codon or an insertion/deletion causing a frameshift, it would be interpreted as a mutation. Deleterious changes in mRNA consensus splice donor and acceptor sites that bracket the exons are also interpreted as mutations. If a missense variant were evolutionarily well conserved and in a critical domain of a protein, it would also usually be diagnosed as a mutation. If no variant were identified by direct sequencing of the coding region, the test should be considered noninformative. In this situation, large genomic rearrangements such as whole exon deletions or insertions must be analyzed by techniques such as Southern blotting or multiplex quantitative PCR of all exons. If no mutations were identified, for certain diseases, immunohistochemistry of formalin-fixed, paraffin-embedded tissue is employed to detect the tumor-specific absence of the protein in question. On rare occasion, a balanced translocation missed by multiplex quantitative PCR is identified with a high-resolution karyotype or by fluorescent in situ hybridization.
Inactivation of specific genes can cause related molecular signatures that can be used to predict individuals who might benefit from genetic testing. A classic example is DNA mismatch repair (MMR) gene mutations in colorectal cancer (CRC), which causes a phenotype of microsatellite instability (MSI) (1,2,3). MSI refers to the contraction or expansion of
short DNA sequences within the tumor DNA that arises due to defective repair of DNA polymerase slippage during replication. These expansions and contractions are directly visible when DNA from a tumor is compared by gel or capillary electrophoresis with DNA from normal epithelium. Defects in the insertion/deletion repair function create a phenotype that is largely believed to be unique to MMR. Tumors lacking MSI are referred to as microsatellite stable (MSS). MSS cancers are generally believed to have greater segmental aneuploidy and gross chromosomal arrangements, perhaps reflecting greater epigenetic abnormalities of DNA methylation and histone modification. It is anticipated that genomic technologies analyzing GI cancers at the levels of genomic DNA, allele-specific expression (allelotyping), mRNA levels, epigenetic modifications, and proteomics will permit recognition of additional molecular signatures of GI cancer subtypes, helping dictate management and therapy. When these molecular signatures of tumor subtypes are defined more precisely, it is anticipated that therapies can be developed that are finely targeted at the pathways affected by specific combinations of mutations in susceptibility genes. With the development of ultra high-throughput DNA sequencing technologies in the coming decade that aim to sequence an entire human genome for $1,000, it is anticipated that “personalized” tumor signatures and signature-directed therapies will begin to emerge.
short DNA sequences within the tumor DNA that arises due to defective repair of DNA polymerase slippage during replication. These expansions and contractions are directly visible when DNA from a tumor is compared by gel or capillary electrophoresis with DNA from normal epithelium. Defects in the insertion/deletion repair function create a phenotype that is largely believed to be unique to MMR. Tumors lacking MSI are referred to as microsatellite stable (MSS). MSS cancers are generally believed to have greater segmental aneuploidy and gross chromosomal arrangements, perhaps reflecting greater epigenetic abnormalities of DNA methylation and histone modification. It is anticipated that genomic technologies analyzing GI cancers at the levels of genomic DNA, allele-specific expression (allelotyping), mRNA levels, epigenetic modifications, and proteomics will permit recognition of additional molecular signatures of GI cancer subtypes, helping dictate management and therapy. When these molecular signatures of tumor subtypes are defined more precisely, it is anticipated that therapies can be developed that are finely targeted at the pathways affected by specific combinations of mutations in susceptibility genes. With the development of ultra high-throughput DNA sequencing technologies in the coming decade that aim to sequence an entire human genome for $1,000, it is anticipated that “personalized” tumor signatures and signature-directed therapies will begin to emerge.
Basic Cancer Genetics
Advances in molecular genetics since 1990 have indicated that cancer is a “disease of DNA” (4,5,6). Mutations in genes passed onto progeny occur in all cells in the body. These mutations are called germline mutations. A mutation present in all cells in a body that is not inherited from a parent is referred to as a de novo mutation. When mutations occur in individual cells, such as during DNA genomic replication or through DNA damage, this type of mutation is referred to as somatic. When a somatic mutation occurs shortly after fertilization and is present in many but not all cells in the body, it is called mosaic.
Germline mutations can be inherited with both chromosome alleles affected (recessive inheritance) or only one chromosome (dominant inheritance). Most GI cancer syndromes have incomplete penetrance. This term refers to the concept that an individual can inherit a mutated gene but not develop any features of the related syndrome in their lifetime. Incomplete penetrance is a common feature of all adult genetic disorders. Expressivity refers to the way in which a gene mutation manifests. For example, a brother and a sister from a family with a Lynch syndrome gene mutation could develop colon or endometrial cancer, respectively. Although they are both affected with Lynch syndrome, the gene mutation expressivity is different for each. Somatic inactivation refers to the situation where a gene either is mutated or has no mutation but has its mRNA transcription repressed by other mechanisms, such as DNA or histone modification or whole gene deletion (6). With reference to GI cancer genetics, almost all syndromes are caused by dominant mutations. Typically, the first mutation is inactivated through a germline inherited or de novo mutation, and the second allele is inactivated through somatic inactivation in individual cells. The classic description of this phenomenon was popularized by Knudson et al. with regard to the retinoblastoma syndrome (4). Somatic mutation and inactivation is believed to play a widespread role in many cancers that normally appear to be sporadic (i.e., without a family history). For example, while germline mutations of the MLH1 gene cause Lynch syndrome, somatic inactivation of MLH1 in both alleles causes an estimated 7.5% of sporadic CRC (7). Therefore, the same gene can be inactivated in hereditary and sporadic CRC, but through different mechanisms.
The three major classes of genes mutated are oncogenes, tumor-suppressor genes, and genes involved in the maintenance of genetic stability. For an in-depth discussion of this topic, the reader is referred to the excellent texts The Genetic Basis of Human Cancer (8) and The Biology of Cancer (6). Briefly, oncogenes and tumor suppressors are mutated genes that directly regulate the growth of cancer cells by inhibiting their growth and progression through the cell cycle or by promoting programmed cell death (apoptosis). Classically, oncogenes are “activated.” A prime example is a somatically acquired missense mutation causing a constitutively active form of KRAS that inappropriately transduces growth-promoting signals commonly in colorectal and pancreatic cancers (7). Tumor-suppressor genes prevent cell cycle progression. Classic examples are SMAD4 (DPC4) inactivation through somatic deletion in pancreatic cancer and APC inactivating mutations in CRC (7,8).
Genes involved in the maintenance of genomic stability include the MMR genes. When these genes are inactivated, somatic mutation rates increase, with the subsequent appearance of oncogene and tumor-suppressor gene mutations. However, there can be some overlap in these categories. For example, the MMR genes play roles both in maintaining genomic integrity and in initiating apoptosis in response to DNA damage (6). The pathogenesis of CRC and other GI neoplasms results from a series of discrete genetic changes that include mutational inactivation of tumor-suppressor genes and activation of oncogenes (reviewed in 7,8). Briefly, mutations in an estimated 5 to 11 genes (9,10,11) are believed to be required for the development of a malignant tumor. These mutations follow a progression from normal epithelium to benign tumors and eventually to cancer by accumulating genetic alterations. Early mutations that confer a selective growth advantage lead to clonal proliferation and accumulation of additional mutations. The order of these mutations may be important because later genetic changes such as mutated TP53 are not sufficient to initiate tumor development as singular early events. In addition, the activation of oncogenes in adult animals can have different consequences than activation during embryogenesis (12). In individuals with genome stability-maintenance genes such as MMR, oncogene and tumor-suppressor mutations accumulate at a faster rate. Recent studies suggest that oncogenes and tumor-suppressor mutations have particularly deleterious consequences when they occur in a small number of cancer-initiating cells with properties of self-renewal analogous to stem cells (13,14).
Colorectal Cancer
CRC is the second leading cause of cancer death in the United States (15). Approximately 10% to 15% of adults in the United States have a family history of CRC in a first-degree relative (16,17). A large proportion of CRC is preventable if detected early, usually with colonoscopic screening (18). Early detection is particularly important in high-risk subjects. Molecular genetics has made a significant impact in decreasing CRC mortality through the identification of genetic syndromes. Therefore, it is important to understand and recognize these syndromes. Since 1990, there have been significant advances in the understanding of the genes that underlie hereditary susceptibility to CRC. It is now well established that mutations in DNA repair genes (MLH1, MSH2, MSH6, MYH, BLM) and genes
involved in signal transduction (APC, LBK1, STK11, SMAD4) underlie a significant percentage of hereditary CRC susceptibility. This section addresses the most important CRC genetic syndromes.
involved in signal transduction (APC, LBK1, STK11, SMAD4) underlie a significant percentage of hereditary CRC susceptibility. This section addresses the most important CRC genetic syndromes.
Lynch Syndrome and DNA Mismatch Repair
The phenotypic spectrum of Lynch syndrome includes cancer of the colorectum, endometrium, stomach, upper urinary tract, small bowel, ovary, and bile ducts. Although usually recognized by clinical criteria (e.g., the revised Bethesda guidelines) (19), colorectal adenocarcinoma arising in Lynch syndrome may often have several recognizable histopathological features, including a poorly differentiated, mucinous appearance, characteristic lymphocytic infiltrate, histologic heterogeneity, and signet ring features (20,21,22). Clinically, approximately two-thirds of colon cancers are right sided, compared to approximately one-third in sporadic cancer. Linkage analyses originally led to the identification of MLH1 and MSH2 mutations in highly penetrant Lynch syndrome pedigrees (23,24). These genes are estimated to cause >90% of Lynch syndrome. Subsequently, identifiable MSH6 mutations have been shown to underlie less penetrant aggregations of CRC (25,26,27). In addition, rare atypical Lynch syndrome pedigrees with identifiable PMS2 or MLH3 mutations have also been described (28,29,30). An algorithm (31) for the clinical evaluation of Lynch syndrome is summarized in Figure 3.1.
Patients with Lynch syndrome mutations have significantly improved survival with intensive cancer surveillance, and family members who do not inherit a mutation are spared the discomfort, expense, and iatrogenic complications of cancer surveillance (32,33,34,35). Because the clinical management and prognosis of mutation versus nonmutation carriers differ so drastically, it is imperative that deleterious mutations be identified, and their clinical impact correctly interpreted. Overall, societal health care costs are decreased with Lynch syndrome diagnosis and subsequent management if the costs of subsequent hospitalization for metastatic disease are included (32,33,34,35). There is significant variation with regard to age of onset, clinical phenotypes, and tumor spectrum among families with Lynch syndrome mutations. The clinical phenotype ranges from families satisfying the highly specific Amsterdam criteria (36) to familial CRC (defined as proband plus either one affected first-degree relative or two affected second-degree relatives), isolated early onset disease, and even sporadic CRC (37). The wide range of clinical phenotypes complicates genetic counseling, and the implementation of individualized medical-surgical management for patients and their relatives carrying these mutations. There is no consensus on how to classify them as deleterious mutations or benign polymorphisms. They are often referred to as variants of uncertain significance. Part of the large variation in the MLH1/MSH2 mutant cancer susceptibility is due to the frequent occurrence of missense mutations. Including all types of mutations (e.g., truncating, genomic deletion, duplication, rearrangement), 24% of hereditary nonpolyposis colon cancer (HNPCC) mutations cited in the International Society for Gastrointestinal Hereditary Tumor database are missense. Missense mutations cause a broad range of clinical phenotypes. Although some cause complete loss of protein function and result in a phenotype similar to mutations that truncate the protein prematurely, many create proteins that retain partial function. There is widespread agreement in the cancer genetics community that correct interpretation of the clinical significance of specific missense mutations is extremely challenging and is a limiting factor in the medical management of the families involved.
Lynch Syndrome Mutation Risk Prediction Clinical Models
The revised Bethesda guidelines were developed to identify screening criteria to identify subjects who are at high risk of having a germline MMR gene mutation (36). More recently, multivariate logistic regression risk models using personal and family medical history have been created to predict more precisely the probability that an individual carries a Lynch syndrome mutation. The results of MSI/IHC studies can also be included with the other information to assess risk. Barnetson et al. (38) and Chen et al. (39) independently developed and validated logistic regression models that predict an individual’s risk of carrying a mutation in MLH1/MSH2/MSH6 (referred to here, respectively, as Barnetson and MMRPro models). Balmana et al. similarly developed a logistic regression model to predict risk of carrying a mutation in MLH1/MSH2 (PREMM1,2) (40). These models, which employ either logistic regression or Bayesian analytic approaches to predict risk, outperform the revised Bethesda guidelines in accuracy of identifying MMR mutation high-risk individuals. At present, it has not been well defined if one model is preferred over the others, and future studies will be required to resolve this issue.
DNA Mismatch Repair and Mechanisms of Cancer Prevention
More than 1 billion years old, MMR plays critical roles in the maintenance of genomic stability in prokaryotes, simple eukaryotes, and metazoan organisms such as humans and rodents (41,42,43). Interest in DNA MMR and its mechanisms of action exploded in the early 1990s with the observation that germline mutations in MMR genes cause HNPCC (23,24,44,45). The biology of MMR has been extensively characterized in model organisms, and these studies have given tremendous insights into the function of specific proteins in humans. The model organisms in which MMR has been most extensively studied are Escherichia coli and Saccharomyces cerevisiae. Other systems in which MMR genes have been rigorously characterized include Caenorhabditis elegans and Arabidopsis thaliana. The precise mechanistic functions that each MMR protein performs in these model organisms, as well as humans and mice, has been covered in detail in numerous excellent reviews (43,46,47,48,49,50,51,52,53). Therefore, this subject is summarized only succinctly here, focusing on mechanisms most likely to be relevant to the discussion of tissue-specific carcinogenesis.
Briefly, there are nine mammalian MMR genes (MLH1, MLH3, PMS1-2, MSH2-6) (43,46). The MMR proteins interact with each other to create a combinatorial code of complexes that mediate distinct functions. The mammalian E. coli MutS homologues (MSH proteins) are believed to directly contact double-stranded DNA, scanning along the genomic DNA for mismatches analogous to a “sliding clamp” until they encounter a base pair containing a mismatch (54,55). The MSH proteins interact with multiple proteins including the mammalian E. coli MutL homologues (MLH) and yeast postmeiotic segregation (PMS) homologue proteins (which have significant amino acid identify and structural similarity to the MLH proteins), as well as RPA, EXO1, RFC, possibly HMGB1, and other less well-characterized proteins (reviewed in 43,46; also see 56,57,58, which discuss this topic in excellent detail). With respect to the mutator function, the MSH2-MSH6 heterodimer is believed to primarily repair single-base substitutions and 1 base pair insertion-deletion mutations, while MSH2-MSH3 is
believed primarily to repair 1 to 4 base pair insertion-deletion mutations (43,46). The E. coli MLH and yeast PMS proteins interact with heterodimers of MSH proteins to help catalyze their different functions. MLH1-PMS2 is the primary MutL complex that interacts with both MSH2/6 and MSH3 complexes in mechanisms believed to be relevant to cancer prevention. Recent studies suggest that mammalian MLH1-MLH3 also contributes to some of these processes, but in all mechanisms tested to a lesser degree than MLH1-PMS2 (29,30,59,60). MLH1-PMS1 clearly exists in mammalian cells, but it currently has no clearly defined role in processes relevant to cancer susceptibility (61,62). MSH4 and MSH5 are believed to play roles exclusively in meiosis and are currently not believed to contribute to mechanisms of cancer prevention (63).
believed primarily to repair 1 to 4 base pair insertion-deletion mutations (43,46). The E. coli MLH and yeast PMS proteins interact with heterodimers of MSH proteins to help catalyze their different functions. MLH1-PMS2 is the primary MutL complex that interacts with both MSH2/6 and MSH3 complexes in mechanisms believed to be relevant to cancer prevention. Recent studies suggest that mammalian MLH1-MLH3 also contributes to some of these processes, but in all mechanisms tested to a lesser degree than MLH1-PMS2 (29,30,59,60). MLH1-PMS1 clearly exists in mammalian cells, but it currently has no clearly defined role in processes relevant to cancer susceptibility (61,62). MSH4 and MSH5 are believed to play roles exclusively in meiosis and are currently not believed to contribute to mechanisms of cancer prevention (63).
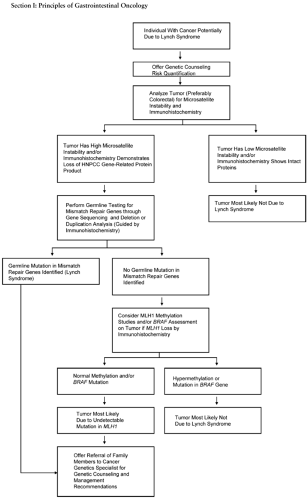 FIGURE 3.1. Algorithm for molecular diagnosis of Lynch syndrome. A consensus approach to the molecular diagnosis of Lynch syndrome in at-risk individuals. Individuals to be evaluated have a thorough personal medical and family history and physical exam. Revised Bethesda guidelines and risk prediction models quantify the likelihood of identifying a mutation in the genes underlying the Lynch syndrome. Molecular analyses are performed on tumor tissue, if available. The molecular phenotype of the tumor guides subsequent genetic testing. Source: Adapted from ref. 257. |
A mutator phenotype causing increased mutation rates for single-base substitution and insertion-deletion mismatches have traditionally been believed to be the main functions in MMR cancer prevention. Defects in the insertion-deletion repair function create a phenotype that is largely believed to be unique to MMR. Because this function was originally characterized on short repetitive DNA microsatellite sequences, it is often referred to as MSI (1,2,3). MSI is divided into MSI-Low and MSI-High (MSI-H) subtypes, depending on the percentage of microsatellite markers that are mutated, but only the MSI-High subtype is clearly associated with defective MMR. Tumors with no MSI are referred to as MSS. (Consensus thinking on the topic of MSI was recently covered in detail by Umar et al. [19].) The precise mechanisms whereby MSI causes cancer are hypothesized to include a genomewide increased mutation rate that causes mutations not only in microsatellites, but also in exonic coding sequences of genes important in cancer suppression. MSI mutation rates in these short repetitive sequences in MLH1- or MSH2-deficient cells are estimated to be more than 100 times higher than that seen in MMR-proficient cells and used to test directly whether MMR deficiency is involved in specific tumor types. Almost all described MLH1/MSH2 mutations cause MSI-H tumors, although recently MSS MLH1 nonsynonymous amino acid substitutions causing deleterious mutations have been described (64,65). Current thinking is that MMR mutations are recessive (i.e., require two hits under the Knudson hypothesis) before cells become susceptible to cancer. However, it has been suggested that cells from subjects carrying heterozygous germline mutations may have detectable MSI using a more sensitive assay referred to as single-molecule or small pool PCR (66,67) that can detect approximately 5- to 10-fold elevations in MSI rates. These new findings are intriguing but require further investigation before the role of MMR haploinsufficiency in cancer susceptibility is clearly established.
In addition to its essential roles in insertion-deletion and single-base substitution repair, MMR proteins also participate in additional mechanisms that could contribute to carcinogenesis, most notably initiation of apoptosis in response to DNA damage (54,68). Recent studies using mouse models of single amino acid substitutions causing separation of function mutations have clearly demonstrated that decreased apoptosis plays an important role in MMR-deficient tumorigenesis (69,70). The precise mechanism by which MMR contributes to the initiation of programmed cell death remains unclear, with both futile cycles of repair causing high levels of double-strand breaks and direct signaling proposed (71,72). MMR mutant cells fail to activate a p73-dependent cell death activation pathway (73), which may explain the absence of a critical requirement for p53-mediated apoptosis (74). This failure impairs cell cycle and DNA damage checkpoint recognition (43).
In summary, the current models suggest that MMR mutations cause cancer primarily through the contribution of both mechanisms: (a) cells acquire mutations in components of critical tumor-suppressor gene pathways that allow them to proliferate, and (b) cells do not initiate apoptosis appropriately. It is unclear whether these mechanisms operate sequentially or concurrently in different cell types. MMR proteins also contribute to suppress homoeologous recombination (71,75), which could in principle contribute as an important mechanism of carcinogenesis. However, the contribution of increased rates of homoeologous recombination from MMR mutations to carcinogenesis is not well defined at this point in time.
Genetic Modifiers of HNPCC
HNPCC is known to have reduced penetrance and variable expressivity. Identifying the major determinants of this phenotypic heterogeneity is critical in tailoring cancer prevention strategies for individual high-risk patients, for example, which families require intensive surveillance for ovarian cancer, and which families can be spared the expense, iatrogenic complications, and inconvenience of this surveillance. Marked interfamily variation in risk of HNPCC spectrum cancer is clearly documented (76,77,78,79,80,81,82,83). The variance is in both (a) age of presentation of CRC in HNPCC, and (b) the distribution of different tumor types that develops in different HNPCC families (76,77,78,79,80,81,82,83). However, the reasons for these phenotypic variances are poorly understood. CRC presentation ranges from the early 20s to no disease in the early 90s for germline mutation carriers (82,83,84). Three population-based studies from Europe and the United States (82,83,84) have studied estimated lifetime CRC penetrance in HNPCC. A meta-analysis of these studies estimates lifetime CRC risk as approximating 74% for males and 39% for females (79).
Variants in the Cyclin D1 gene have been proposed to be a genetic modifier of HNPCC penetrance. Cyclin D1 reaches maximal activity during the G1 phase, in which it plays an important role in the transition from the G1 phase to the S phase of the cell cycle. The Cyclin D1 gene has a G to A variant at codon 242 in exon 4 that increases alternate splicing, creating a protein with significantly increased protein stability. HNPCC patients who are homozygous or heterozygous for the mutant allele developed CRC an average of 11 years earlier than patients who were homozygous for the normal alleles (85). However, this putative association has only been reported in one group of patients, and no validation in additional cohorts of HNPCC patients has been performed. Thus, it is unclear whether this putative modifier effect on HNPCC CRC onset is a true or chance association.
NAT2 is a highly polymorphic isozyme of N-acetyltransferase and is found in a variety of tissues, including the colorectal mucosa. It catalyzes the metabolism of xenobiotics and carcinogens by transferring an acetyl group to these agents, and has been suggested as a potential modifier of CRC risk. Frazier et al. (86) found HNPCC patients heterozygous for the variant allele NAT2*7 after adjustment for the NAT2 mutant loci NAT2*5, and NAT2*6 had a significantly higher risk of CRC (hazard ratio, 2.96; P <0.012) than individuals homozygous for the wild-type allele. However, without this adjustment, NAT2*7 allele carriers were not at increased CRC risk. Therefore, it is unclear whether this modifier effect is true or was ascertained only through multiple hypothesis testing.
Recently, this relationship has been clarified by several demonstrations that cigarette smoking selectively increases the subset of CRC that manifests MSI-High (77,87). The environmental component of this interaction can be incorporated through a quantitative assessment of lifetime smoking exposure, and quantitative estimates of smoking risk performed (79). However, it is likely that genetic factors impact the effect of smoking as well. For example, genetic factors have been described that significantly influence the ability of individuals to quit smoking (88,89), which likely has an important impact on lifetime smoking exposure.
Management
Individuals with known Lynch syndrome MLH1/MSH2 mutations have colonoscopic surveillance every 1 to 2 years (31). Surveillance should start at ages 20 to 25. MSH6 mutation carrier surveillance is not as well defined, but age 30 is often used for these patients (31). In women with Lynch syndrome mutations, there are no adequately powered studies that evaluated whether endometrial cancer screening improves survival. Nonetheless, as a high-risk population, endometrial cancer screening in these patients is reasonable. MSH6 mutation carriers in particular may have greater endometrial cancer than CRC risk. Transvaginal ultrasound has been used, but there is a high false-positive rate (31). Endometrial biopsy is used in women who are symptomatic, such as presenting with pelvic pain and/or irregular pre- or postmenopausal vaginal bleeding. Endometrial biopsy beginning at ages 30 to 35 can be used every 1 to 2 years for surveillance. The absolute risk of Lynch syndrome carriers for other sites (e.g., ovary, brain, renal, stomach, bladder, biliary tract) is greater than the general population but is clearly less than colorectum and endometrium. Screening for these sites is therefore not well defined. Many clinicians will perform annual urinalysis with cytology and transvaginal ultrasound for ovarian cancer every 1 to 2 years as a reasonable surveillance strategy. A baseline upper endoscopy should be performed, but the optimal subsequent screening interval has yet to be established. An important general rule is that when families have a particular tumor type that is prominent, increased surveillance should be instituted for the tissue site. So, families that have gastric cancer should have endoscopy at 1- to 2-year intervals.
In terms of surgical management, prophylactic colectomy before the identification of colon cancer is typically not performed. However, once a first malignancy is identified, a subtotal colectomy with retention of the rectum (which preserves fecal continence) is performed. This management is in contrast to the segmental colectomy typically performed for sporadic colon cancer. After surgery, the rectum can be monitored with proctoscopy, sigmoidoscopy, or limited colonoscopy. Because there are no studies formally comparing subtotal colectomy versus segmental colectomy with frequent colonoscopic surveillance, patient preference is important to integrate into the surgical management strategy. For ovarian and endometrial cancer surgical management, a retrospective study of prophylactic bilateral oophorectomy and hysterectomy in 315 women with Lynch syndrome showed that after 10 years of follow-up, 0% of uterine/endometrial cancer developed in the women who had surgery, but 5.5% of the women who did not have surgery developed ovarian cancer, and 33% of these women developed endometrial cancer (90). Based on these data, prophylactic bilateral oophorectomy and hysterectomy is considered a reasonable option for women with Lynch syndrome. Women who do consider this option typically wait until after childbearing. Often, subtotal colectomy, oophorectomy, and hysterectomy are performed at the same time.
In addition to Lynch syndrome, there are at least three other relatively common forms of hereditary predisposition to colorectal neoplasia: familial adenomatous polyposis (FAP), attenuated FAP, and Peutz-Jeghers syndrome (PJS).
Familial Adenomatous Polyposis
Originally described in the late 19th century, FAP is a rare (1 in 7,000–8,000) syndrome characterized by presentation with hundreds or even thousands of colonic polyps in the late teens or early 20s and progression to colon cancer in virtually every case. A minimum of 100 polyps is generally necessary to make the diagnosis, whereas HNPCC families rarely have more than 50 polyps. Median age of adenomas is 16 (91), with 95% affected by 35 (92). Onset of CRC is 100% by age 40 (93), with average age of 36 (91). Benign hamartomas are seen in the fundus and body in about 50% of patients, and duodenal polyps in about 90%, with a 4% to 12% risk for duodenal/ampullary carcinoma. There is also an increased risk for small bowel cancer and thyroid cancer (follicular or papillary), mostly in women. Children are at risk for hepatoblastoma, and CNS tumors (medulloblastomas) can be seen in the Turcot variant. Gardner syndrome (94) is characterized by cutaneous soft tissue tumors; lipomas; fibromas; epidermoid/sebaceous cysts on legs, face, scalp, and arms; osteomas (skull and mandible); supernumerary teeth; congenital hypertrophy of the retinal pigment epithelium (CHRPE); and desmoid tumors and mesenteric fibromatosis (95,96,97,98,99). There is an association between FAP and juvenile nasopharyngeal angiofibroma, which is 25-fold more prevalent in men with FAP than in the general population (100,101,102,103,104,105,106,107).
An Israeli study documented extracolonic manifestations among 38 of 50 FAP patients, or 76%. Two of the 50 patients died, 1 from a mesenteric desmoid tumor and 1 of mesenteric malignant fibrous histiocytoma (108).
APC
The APC gene (5q21) encodes a large protein that acts in the Wnt pathway via several domains: to regulate cell cycle and apoptosis, to stabilize the cytoskeleton, and to mediate intercellular adhesion (109). The protein product of the APC gene was shown to associate with proteins (catenins) that bind to a cell surface molecule (cadherin), which is essential for cellular adhesion. These studies suggest that the APC protein may be a vital communications link between the cell surface and the microtubules necessary for cell division (110,111,112). An important region binds with ß-catenin, inhibiting it from interacting with T-cell factor/lymphoid enhancer factor (Tcf/Lef) to form a proliferative DNA transcription complex (113,114). About 30% of APC mutations are de novo, and 98% are truncating (115). Interstitial deletions of 5q22, which includes the APC gene, have been reported in individuals with adenomatous polyposis and mental retardation.
Severe polyposis is associated with APC mutations in codons 1250 to 1464, sparser polyps with mutations in codons 213 to 1249 and 1465 to 1597, CHRPE with mutations in codons 311 to 1444, and Gardner syndrome with mutations in codons 1395 to 1578. There is no clear relationship for duodenal, periampullar, and gastric adenomas (91,116,117,118,119,120,121,122,123,124,125).
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


