CHAPTER 49 Gastric Secretion
The stomach is an active reservoir that stores, grinds, and slowly dispenses partially digested food to the intestine for further digestion and absorption. Its main secretory function is the production of hydrochloric acid. Gastric acid secretion is present on the first day of life and increases as infants become more mature.1 By two years of age, acid secretion is similar to that of adults, when corrected for body weight.2 Most studies indicate that the rate of acid secretion changes little after the second decade of life unless there is coexisting disease of the acid-secreting glandular mucosa such as infection with Helicobacter pylori (HP) or atrophic gastritis.3–5
Acid facilitates the digestion of protein by converting the proenzyme pepsinogen to the active proteolytic enzyme pepsin. It also facilitates the absorption of iron, calcium, vitamin B12, and certain medications (e.g., thyroxin) as well as prevents bacterial overgrowth, enteric infection, and possibly community-acquired pneumonia.6–18
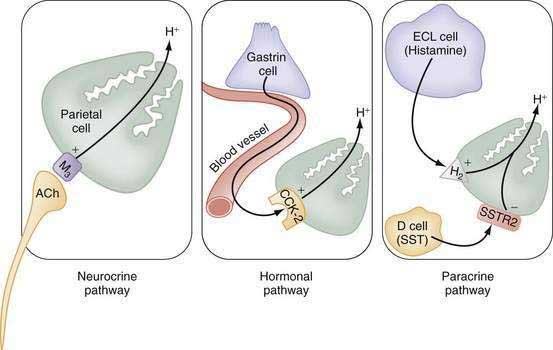
The stomach also secretes lipase, intrinsic factor, electrolytes (e.g., HCO3−, K+, and Cl−), and mucins in addition to a variety of neurocrine, paracrine, and hormonal agents (Fig. 49-1). Neurocrine agents are released from nerve terminals and reach their targets via synaptic diffusion (e.g., acetylcholine [ACh], gastrin-releasing peptide [GRP], and vasoactive intestinal peptide [VIP]). Paracrine agents are released in proximity to their targets and reach them via diffusion (e.g., histamine and somatostatin). Hormones are released into the circulation and reach their targets via the bloodstream (e.g., gastrin).
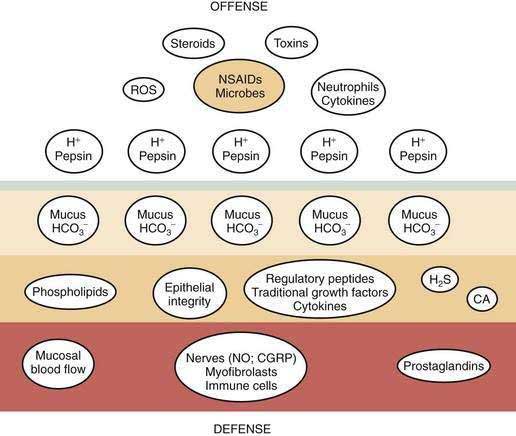
Gastric mucosal integrity depends on a delicate balance between secretion of aggressive (e.g., acid and pepsin) and defensive (e.g. bicarbonate and mucin) substances (Fig. 49-2).19 When mucosal defense mechanisms are overwhelmed, ulceration may occur. In order to reap the benefits of acid without untoward effects, gastric exocrine and endocrine secretion must be precisely regulated. This is accomplished by a highly coordinated interaction among a multitude of neural, paracrine, and hormonal pathways.
FUNCTIONAL ANATOMY
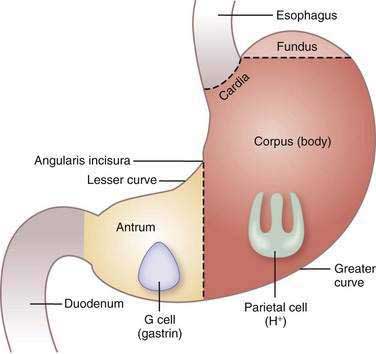
The stomach consists of three anatomic (fundus, corpus or body, and antrum) and two functional (oxyntic and pyloric gland) areas (Fig. 49-3). The oxyntic gland area, the hallmark of which is the oxyntic (oxys, Greek for acid) or parietal cell, comprises 80% of the organ (fundus and corpus). The pyloric gland area, the hallmark of which is the G or gastrin cell, comprises 20% of the organ (antrum). The human stomach contains approximately 1 × 109 parietal cells and 9 × 106 gastrin cells.20 There is debate as to whether the cardia, a transition zone of 0 to 9 mm between the squamous mucosa of the esophagus and the oxyntic mucosa of the stomach, exists as a normal anatomic structure or develops as a result of abnormal reflux. Autopsy and endoscopic studies suggest that cardiac mucosa is absent in more than 50% of the general population.21 Gastric anatomy is discussed in greater detail in Chapter 47.
The glandular area is organized in vertical tubular units that consist of an apical pit region, an isthmus, and the actual gland region that forms the lower part of the unit; the latter consists of a neck and a base (Fig. 49-4). The progenitor cell of the gastric unit, located in the isthmus, gives rise to all gastric epithelial cells. In the oxyntic gland area, the mucus-producing pit cells migrate upward from the progenitor cell toward the gastric lumen. Acid-secreting parietal cells migrate downward to the middle and lower regions of the gland; those at the bases are less active acid secretors. The turnover time for parietal cells is 54 days in mice and 164 days in rats.20 Chief (zymogenic) cells predominate at the base and secrete pepsinogen and leptin22; the latter is also present in parietal cells.23 Several distinct neuroendocrine cell types are contained within the gland but only some of their products have been assigned physiologic functions (see Chapter 1). The cells include enterochromaffin (EC) cells (atrial natriuretic peptide [ANP], serotonin, and adrenomedullin), enterochromaffin-like (ECL) cells (histamine), D cells (somatostatin and amylin), and A-like or Gr cells (ghrelin and obestatin).24–29 ECL cells constitute 66% of the neuroendocrine cell population in rats and 30% in humans. Somatostatin-containing D cells possess neural-like cytoplasmic processes that terminate in the vicinity of parietal and ECL cells (see Fig. 49-4). The functional correlate of this anatomic coupling is a tonic paracrine restraint exerted by somatostatin on acid secretion directly as well as indirectly by inhibiting histamine secretion (Fig. 49-5).30–32
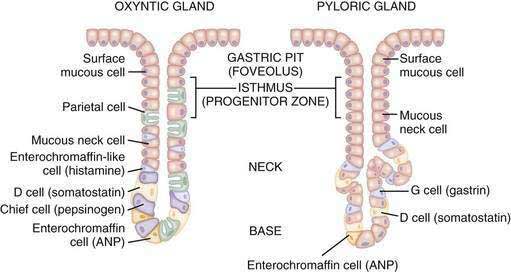
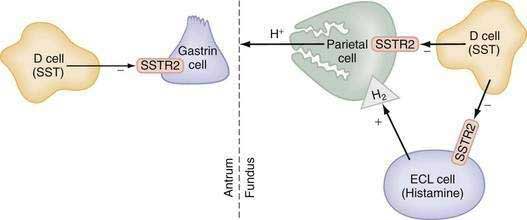
Somatostatin-containing D cells are also present in the pyloric gland area (see Figs. 49-4 and 49-5); in this region they exert a tonic paracrine restraint on gastrin secretion from G cells.33,34 The pyloric gland also contains EC cells (ANP and serotonin), A-like or Gr cells (ghrelin and obestatin), and endocrine cells containing orexin.26,35,36
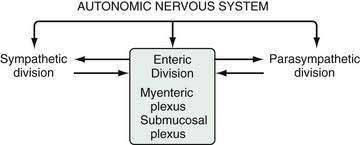
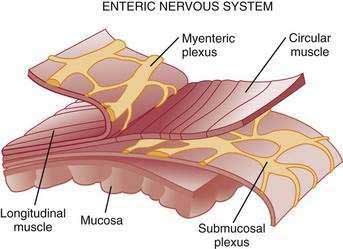
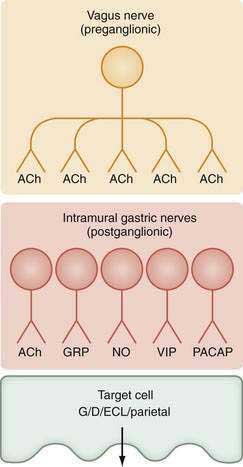
The stomach is innervated by a neural network, the enteric nervous system (ENS), that contains intrinsic neurons, that is, neurons whose cell bodies are contained within the gastric wall (Figs. 49-6 to 49-8). The ENS, the third division of the autonomic nervous system (the other two being the sympathetic and parasympathetic), is often referred to as the “little brain” because it contains as many neurons as the spinal cord, approximately 106, and can function autonomous of central input (see Fig. 49-6).37 Nevertheless, the ENS receives input from and sends input to the central nervous system via sympathetic and parasympathetic neurons. In rats and guinea pigs, most of the intrinsic neural innervation of the stomach originates in the myenteric plexus, located between the circular and longitudinal muscle layers; the submucosal plexus in these species, adjacent to the mucosal layer, contains only a small number of neurons. Humans, in contrast, have a clearly defined submucosal plexus that regulates gastric secretion and contains a variety of neurotransmitters (see Figs. 49-7 and 49-8). It should be noted that the vagus nerve is predominantly afferent, containing 80% to 90% afferent fibers and 10% to 20% efferent fibers. The efferent fibers arise from the dorsal motor nucleus of the brainstem. They are preganglionic and do not directly innervate parietal or neuroendocrine cells but rather synapse with postganglionic neurons of the ENS. The postganglionic neurons contain a variety of transmitters including ACh, GRP, nitric oxide (NO), VIP, and pituitary adenylate cyclase–activating polypeptide (PACAP) (see Fig. 49-8).38 In the stomach, afferent nerve fibers containing calcitonin gene–related peptide (CGRP) are of extrinsic origin, that is, the cell bodies are located outside the stomach wall.39 Postganglionic neurons of the ENS regulate acid secretion directly, as is the case for ACh, and/or indirectly by modulating the secretion of gastrin from G cells, somatostatin from D cells, and possibly histamine from ECL cells (see Fig. 49-8).
ACID SECRETION: PARACRINE, HORMONAL, NEURAL, AND INTRACELLULAR REGULATION
Parietal cells secrete hydrochloric acid at a concentration of approximately 160 mM or pH 0.8. Acid is thought to gain access to the lumen via channels in the mucus layer created by the relatively high intraglandular hydrostatic pressures generated during secretion, about 17 mm Hg.40
Acid facilitates the digestion of protein and absorption of iron, calcium, and vitamin B12 as well as prevents bacterial overgrowth, enteric infection, and possibly community acquired pneumonia.6–14,18,41 However, when levels of acid (and pepsin) overwhelm mucosal defense mechanisms, ulcers occur. To prevent such damage, gastric acid must be precisely regulated and produced according to need. This is accomplished by a highly coordinated interaction among a number of neural, hormonal, and paracrine pathways. These pathways can be activated directly by stimuli originating in the brain or reflexively by stimuli originating in the stomach after ingestion of a meal such as mechanical stimulation (e.g., distention) or chemical stimulation (e.g., protein and acid).
The principal stimulants of acid secretion are (1) ACh, released from postganglionic enteric neurons (neurocrine), (2) gastrin, released from antral G cells (hormonal), and (3) histamine, released from oxyntic ECL cells (paracrine) (see Fig. 49-1; Fig. 49-9). These agents interact with specific G protein–binding receptors (M3, CCK-2, and H2, respectively) that are coupled to two major signal transduction pathways: intracellular calcium in the case of gastrin and ACh, and adenylate cyclase or adenosine 3′,5′-cyclic monophosphate (cAMP) in the case of histamine (see Fig. 49-9). There is evidence for potentiation (or synergism) between histamine and either ACh or gastrin, probably as a result of postreceptor interaction between the two signaling pathways.42 The main inhibitor of acid secretion is somatostatin, released from oxyntic and antral D cells (paracrine) (see Figs. 49-1, 49-5, and 49-9). Each of these agents acts directly on the parietal cell as well as indirectly by modulating the secretion of neuroendocrine cells (Fig. 49-10).
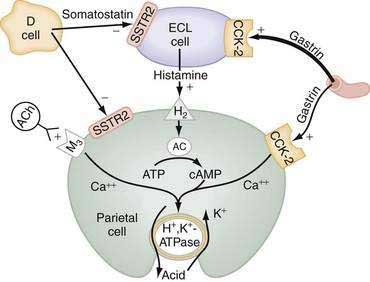
HISTAMINE
Histamine, produced in ECL cells by decarboxylation of l-histidine by histidine decarboxylase (HDC), stimulates the parietal cell directly by binding to H2 receptors coupled to activation of adenylate cyclase and generation of cAMP (see Fig. 49-9).43 Histamine also stimulates acid secretion indirectly by binding to H3 receptors coupled to inhibition of somatostatin release from oxyntic D cells, thus resulting in stimulation of histamine release and acid secretion (see Fig. 49-10).44,45 Gastrin, PACAP, VIP, and ghrelin stimulate, whereas somatostatin, CGRP, prostaglandins, peptide YY (PYY), and galanin inhibit histamine secretion.46,47 As discussed following, gastrin also exerts a direct proliferative effect on ECL cells. ACh has no direct effect on histamine secretion.48–50
GASTRIN
Gastrin, the main stimulant of acid secretion during meal ingestion, is produced in G cells of the gastric antrum and, in much lower and variable amounts, in the proximal small intestine, colon, and pancreas. Gastrin is synthesized as a large precursor molecule of 101 amino acids, which is converted to progastrin (80 amino acids) by cleavage of the N-terminal signal peptide. Progastrin is further processed to yield peptides with C-terminal glycine, that is, G34gly and G17gly. The final processing step involves amidation to yield G34amide and G17amide. The plasma half-life of G34amide is 30 minutes and that of G17amide is three to seven minutes; they are metabolized primarily by the kidney and, in addition, by the intestine and liver.51,52 Consequently, most gastrin in the circulation during fasting is G34, whereas after a meal it is G17. In patients with renal insuffiency as well as massive small bowel resection, fasting blood levels of G17 and G34 are elevated.53,54 It should be noted that the commercially available test substance pentagastrin is not a naturally occurring peptide but rather is a manufactured analog that contains the biologically active C-terminus sequence Trp-Met-Asp-Phe-NH2.
Gastrin and cholecystokinin (CCK) belong to the same family of peptides and possess an identical carboxyl-terminal pentapeptide sequence (-Gly-Trp-Met-Asp-Phe-NH2). Two main classes of gastrin/CCK receptors have been characterized: CCK1 (formerly CCK-A) and CCK-2 (formerly CCKB or CCKB/gastrin). CCK1 receptors are specific for CCK, whereas CCK-2 receptors recognize both CCK and gastrin with high affinity. Gastrin, acting via CCK-2 receptors that activate phospholipase C to induce release of intracellular calcium, stimulates the parietal cell directly and, more importantly, indirectly by releasing histamine from ECL cells (see Figs. 49-9 and 49-10).55,56 Gastrin regulates the secretion and synthesis of histamine in a biphasic manner. The first phase involves release of stored histamine. The second phase relates to the replenishment of histamine stores and involves an increase in HDC activity followed by an increase in HDC gene transcription.57 H2 receptor, HDC, and CCK-2 receptor knockout mice manifest decreased acid secretion, especially in response to gastrin.58–60
Although amidated gastrins had been thought to be the only forms with biological activity, glycine-extended gastrins may regulate the capacity of the parietal cell to respond to secretagogues, release histamine from ECL cells, and stimulate proliferation of colonic mucosa and colorectal cancers.61,62 ACh, GRP, secretin, β2/β3-adrenergic agonists, calcium, protein, and alcoholic beverages produced by fermentation stimulate, whereas somatostatin, galanin, and adenosine inhibit gastrin secretion. In addition, at least two negative-feedback pathways, mediated via release of somatostatin, regulate gastrin secretion. The first is activated by luminal acidity and involves sensory CGRP neurons (see Fig. 49-10). Low intragastric pH (high intragastric acidity) activates CGRP neurons that, via an axon reflex, stimulate somatostatin and thus inhibit gastrin secretion.63–65 Conversely, when intragastric pH rises (low intragastric acidity), for example, by administering antisecretory medications such as proton pump inhibitors (PPIs) or by developing gastric atrophy, somatostatin secretion is inhibited and patients develop hypergastrinemia. There is some evidence that bacterial overgrowth induced by hypochlorhydria may also contribute to hypergastrinemia.66 The second negative-feedback pathway involves a paracrine action whereby gastrin directly stimulates somatostatin and thus attenuates its own secretion (see Fig. 49-10).67
Gastrin also functions as a trophic hormone to stimulate mucosal proliferation. CCK-2 receptors have been localized to the progenitor zone in oxyntic glands, and chronic hypergastrinemia induces proliferation of ECL and parietal cells directly as well as indirectly via the autocrine or paracrine action of growth factors such as heparin-binding epidermal growth factor, amphiregulin, transforming growth factor-α, metalloproteinases, and regenerating islet-derived 1.68,69 Rats rendered hypergastrinemic by a PPI demonstrate a five-fold increase in the number of ECL cells and a 1.5-fold increase in the number of parietal cells.70 Gastrin acts directly on ECL cells to induce hyperplasia, dysplasia, and eventually neoplasia (carcinoids).71 In contrast to rodents, humans rarely develop carcinoid tumors in response to hypergastrinemia unless other factors are present such as chronic active gastritis or gastrinoma associated with multiple endocrine neoplasia type 1 (see Chapter 31).72 Because ECL cells contain somatostatin subtype 2 receptors (SSTR2), somatostatin scintigraphy with 111indium-diethylenetriamine pentaacetic acid [111In-DTPA]octreotide is the preferred imaging method to detect carcinoid tumors (see Chapter 31).73,74
ACETYLCHOLINE
ACh, released from postganglionic neurons whose cell bodies are located primarily in the submucosal (Meissner’s) plexus, stimulates the parietal cell directly as well as indirectly by inhibiting somatostatin secretion (see Fig. 49-10). The parietal cell muscarinic receptor is of the M3 subtype.75,76 Like CCK-2 receptors, M3 receptors are coupled to activation of phospholipase C with generation of inositol trisphosphate and release of intracellular calcium (see Fig. 49-9).77 Alcoholic beverages produced by fermentation stimulate gastric acid secretion and the effect may be mediated via activation of M3 receptors.78 ACh also stimulates acid secretion indirectly by activating M2 and M4 receptors on D cells coupled to inhibition of somatostatin secretion, thus removing the tonic restraint exerted by this peptide on gastrin, ECL, and parietal cells (see Fig. 49-10).
SOMATOSTATIN
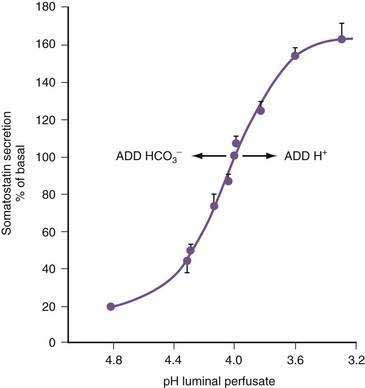
In the stomach, somatostatin cells are closely coupled to their target cells (gastrin cells in the antrum, or parietal and ECL cells in the fundus/body) either directly via cytoplasmic processes or indirectly via the local circulation.28,79 The functional correlate of this anatomic coupling is a tonic restraint exerted by somatostatin on acid secretion from the parietal cell, histamine secretion from the ECL cell, and gastrin secretion from the G cell (see Figs. 49-5 and 49-10).30,31,34,80,81 Removing this restraint (i.e., disinhibition or elimination of the influence of an inhibitor), by activation of cholinergic neurons, is an important physiologic mechanism for stimulating acid secretion (see Fig. 49-10). In the stomach, the actions of somatostatin are mediated primarily via the somatostatin subtype 2 receptor (SSTR2).82–84 Gastrin, GRP, VIP, PACAP, β2/β3-adrenergic agonists, secretin, ANP, adrenomedullin, amylin, adenosine, and CGRP stimulate, whereas ACh and interferon-γ inhibit somatostatin secretion. As mentioned, an increase in luminal acidity acts to attenuate acid secretion via a pathway involving release of somatostatin in the antrum and the fundus. The change in gastric somatostatin secretion can be demonstrated over a range of pH 3 to pH 5, which is within the range observed after ingestion of a meal (Fig. 49-11).85
MISCELLANEOUS PEPTIDES
Ghrelin, the natural ligand for the growth hormone secretagogue receptor, is present in greatest concentrations in gastric oxyntic mucosa and is localized to A-like (or Gr) cells.86,87 Lesser amounts are present in the antrum, small intestine, and colon (see Chapter 1). Plasma ghrelin concentrations increase before meals and decrease postprandially.88 It is postulated that ghrelin triggers premeal hunger and promotes feeding. Its suppression after Roux-en-Y gastric bypass may, in part, contribute to weight loss.89 Most studies report that exogenously administered ghrelin stimulates acid secretion.90,91 The stimulatory effect appears to involve the vagus nerve and histamine release because it is abolished by vagotomy and is associated with an increase in HDC messenger ribonucleic acid (mRNA).92,93
Orexin-A, derived from propro-orexin by post-translational processing, is co-localized with gastrin in human pyloric mucosa.91,94 Intracerebroventricular and peripherally administered orexin-A stimulate gastric acid secretion.95 In rats equipped with gastric fistulas, an orexin receptor 1 antagonist inhibits basal and pentagastrin-stimulated acid secretion, implying that endogenous orexin-A stimulates acid secretion.94,95
ANP, CCK, secretin, neurotensin, glucagon-like peptide 1 (GLP-1), glicentin, oxyntomodulin, peptide YY, adrenomedullin, amylin, glucose-dependent insulinotropic polypeptide (GIP), leptin, epidermal growth factor, and interleukin-1β (IL-1β) inhibit acid secretion. The effect of each, except perhaps for IL-1β, is mediated via release of somatostatin.24–2696 The term enterogastrone has been used to describe the intestinal factor or factors responsible for inhibiting acid secretion in response to nutrients in the intestine. Prime candidates include CCK, secretin, neurotensin, GLP-1, glicentin, and oxyntomodulin because they are present in intestinal mucosa, released into the circulation in response to luminal nutrients, and capable of inhibiting acid secretion at “physiologic” concentrations.97–101 Although it is likely that enterogastrone activity represents the combined influence of several of these peptides, the strongest evidence favors CCK. CCK, produced in I cells in the proximal small intestine, is released by luminal protein and fat. The acid-inhibitory response to intraduodenal fat is abolished by pretreatment with a CCK-1 receptor antagonist in dogs, and the response is blocked in CCK-1 receptor knockout mice as well.102–105
PARIETAL CELL INTRACELLULAR PATHWAYS
In parietal cells, acid secretion is increased by activation of intracellular cAMP- and calcium-dependent signaling pathways that activate downstream protein kinases, ultimately leading to fusion and activation of H+,K+-ATPase (the proton pump) with concomitant activation of luminal membrane conductances for K+ and Cl− (see Fig. 49-9; Fig. 49-12). The H+,K+-ATPase actively pumps out H+ against a tremendous concentration gradient (cell interior pH 7.4 or 40 nM; acid secreted at pH 0.8 or 160 million nM) in exchange for luminal K+. The energy required comes from adenosine triphosphate (ATP) produced by the parietal cell’s extensive mitochondrial network. H+,K+-ATPase consists of an α–subunit that carries out the catalytic and transport function of the enzyme and contains sequences responsible for apical membrane localization106 as well as a β-subunit, which is heavily glycosylated, and protects the enzyme from degradation and is necessary for trafficking to and from the luminal membrane.107
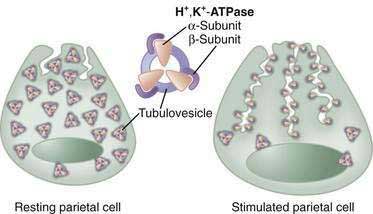
In the resting unstimulated state, H+,K+-ATPase activity is sequestered within cytoplasmic tubulovesicles. On stimulation, there is a dramatic morphologic transformation as these vesicles fuse with the apical plasma membrane, resulting in a 6- to 10-fold increase in the membrane and the formation of the canalicular system (Fig. 49-13). Translocation of the H+,K+-ATPase into the canalicular membrane together with the presence of luminal K+ activates the enzyme.108 On cessation of secretion, the H+,K+-ATPase is retrieved from the apical membrane and the tubulovesicular compartment is reestablished. The precise mechanisms regulating trafficking are not known, but data suggest that it involves actin-based microfilaments, small GTPases, docking/fusion proteins, ezrin, and clathrin.109–111
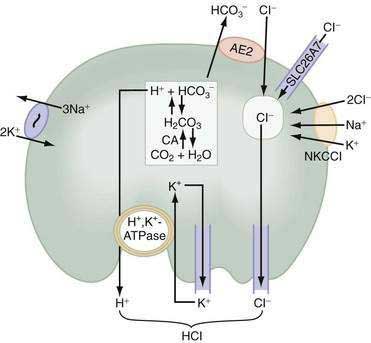
Acid secretion requires not only a functional H+,K+-ATPase but also apical K+ and Cl− channels and basolateral HCO3− and Cl− exchangers. Acid is produced from the hydration of CO2 to form H+ and HCO3−, a reaction catalyzed by cytoplasmic carbonic anhydrase (see Fig. 49-12). Because the H+,K+-ATPase is unable to pump H+ into the lumen without a parallel uptake of K+, sufficient quantities of K+ must be delivered to the lumen. This K+ recycling is accomplished by luminal KCNE2/KCNQ1 and ROMK (KCNJ1) potassium channels. KCNQ1 is a voltage-activated K+ channel, which, when modified by the small regulatory subunit, KCNE2, becomes voltage insensitive, constitutively open, and acid activated.108,112,113 ROMK may be regulated by the cystic fibrosis transmembrane conductance regulator (CFTR).108,112–114 The concentration of K+ in gastric juice (8 to 12 mM) exceeds plasma K+ by two- to four-fold.
For each H+ secreted, an HCO3− ion exits the cell across the basolateral membrane via the anion exchanger 2 (AE2), Slc4a2 (see Fig. 49-12).115 As a result of this HCO3−/Cl− exchange, the pH within the parietal cell remains only slightly alkaline during acid secretion.116 Rapid entry of HCO3− from parietal cells into blood has been referred to as the alkaline tide. Some of this HCO3− may be taken up and secreted by surface epithelial cells.
Concurrently with H+, Cl− is extruded across the luminal membrane via an apical chloride channel, the precise identity of which is not known. The sources of intracellular Cl− are the basolateral anion exchanger 2 (AE2), sodium-2 chloride potassium-cotransporter-1 (NKCC1), and the SLC26A7 channel (see Fig. 49-12).117,118
PROTON PUMP INHIBITORS
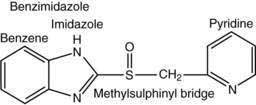
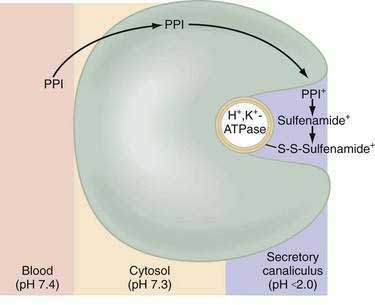
Current PPIs (e.g., omeprazole, lansoprazole, rabeprazole, pantoprazole, and esomeprazole) consist of two heterocyclic moieties, a pyridine and a benzimidazole ring, connected by a methylsulphinyl group (Fig. 49-14). They are weak bases (pKa 4 or 5) that concentrate in acidic spaces within the body that have a pH less than 4. The pKa of a molecule refers to the degree of willingness of the compound to accept or donate a proton and is based on a logarithmic scale such that a compound with a pKa of 5 is 10-fold more basic than a compound with a pKa of 4. When a compound is in an environment with a pH equal to its pKa, half the molecules will be protonated and half will be non-protonated. If a compound with a pKa of 4 is placed in an environment with a pH less than 1, more than 99.9% of the molecules will be protonated. PPIs are membrane permeable in the nonprotonated form and relatively impermeable in the protonated form. In blood (pH 7.4), PPIs are predominantly nonprotonated and thus pass readily into cells (time to reach peak plasma concentration, ≈2 hours; elimination half-life, ≈1 hour), diffuse through the cytoplasm, and then become protonated and trapped, probably as sulfenamides or sulfenic acid, in the acid environment of the secretory canaliculus (Fig. 49-15).119 As a consequence of the basic pKa of PPIs and “ion trapping,” the concentration of PPIs in the secretory canaliculus is 100,000- to 1,000,000-fold higher than in the blood. The sulfenamide rapidly reacts with cysteines on the luminally exposed α-subunit of the H+,K+-ATPase to form a covalent (electrochemical) disulfide bond (see Fig. 49-15).120 Whereas all PPIs bind to cysteine 813, omeprazole also binds to cysteine 892, lansoprazole to cysteine 321, and pantoprazole to cysteine 822. Because only the apical-membrane inserted H+,K+-ATPase is susceptible to blockade by PPIs and an acid environment (pH < 4) is necessary for trapping and converting the PPI to its reactive species, the potency of PPIs is decreased when administered during the basal (fasting) state or when acid secretion is inhibited.121,122 Because most pumps are inserted with breakfast, it is recommended that PPIs be taken 30 minutes to 1 hour before the first meal. If greater inhibition is needed, an additional dose should be taken before dinner. Recovery from inhibition of acid secretion occurs by de novo synthesis of pump protein (54 hours in rat). It has been postulated that reduction of the cysteine disulfide bonds by reducing agents such as glutathione (15 hours in rat) could also play a role.123
INTEGRATED RESPONSE TO A MEAL: INTERPLAY OF NEURAL, PARACRINE, AND HORMONAL PATHWAYS
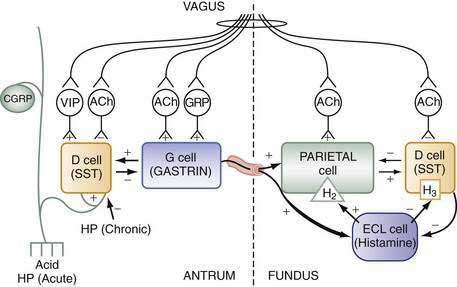
Stimuli originating inside and outside the stomach converge on gastric intramural efferent neurons that are the primary regulators of acid secretion. The effector neurons comprise cholinergic and peptidergic (i.e., GRP and VIP) neurons. Although nitric oxide and PACAP neurons are present in gastric mucosa their precise physiologic roles as regulators of acid secretion are not known. The effector neurons act on target cells directly as well as indirectly by regulating secretion of gastrin, somatostatin, and possibly histamine (see Figs. 49-8 and 49-10).
During the basal state, acid secretion is maintained at an economically low level by the continuous inhibitory restraint exerted by somatostatin on the ECL cell (histamine) and parietal cell (acid) in the fundus/body and on the G cell (gastrin) in the antrum (see Figs. 49-5, 49-9, and 49-10). During ingestion of a meal, maximal secretion may be achieved by removing the inhibitory influence of somatostatin while at the same time directly stimulating acid and gastrin secretion. This is accomplished, in large part, by activation of intramural cholinergic neurons (see Fig. 49-10). The thought, sight, smell, and taste of food contributes up to 50% of total postprandial acid secretion.124,125 Anticipation of a meal activates central neurons whose input is relayed via the vagus nerve to gastric intramural cholinergic neurons in oxyntic as well as pyloric mucosa. The components of the central nervous system include the dorsal motor nucleus of the vagus, the nucleus tractus solitarius, and the hypothalamus. In the fundus/body, ACh, released from cholinergic neurons, stimulates the parietal cell directly, as well as indirectly, by eliminating the inhibitory paracrine influence of somatostatin on parietal and ECL cells.44,126 The resultant increase in histamine stimulates acid secretion directly via H2 receptors on the parietal cell and indirectly via H3 receptors that mediate suppression of somatostatin secretion (see Fig. 49-10).44,127 Thus, histamine, acting via H3 receptors, amplifies the ability of secretagogues to stimulate acid secretion by suppressing somatostatin secretion. The net effect of cholinergic neurons is suppression of all paracrine inhibitory influence (i.e., somatostatin) and enhancement of paracrine stimulatory influences (i.e., histamine acting via H2 receptors) on parietal cells. There is some evidence that PACAP, a member of the glucagon/VIP superfamily of regulatory peptides, may participate in the regulation of acid secretion, but its precise physiologic role is not known.26,128,129 PACAP is present in gastric mucosal nerves and capable of releasing histamine from ECL cells and somatostatin from D cells. The net effect of exogenous PACAP on acid secretion has been reported to be either stimulation or inhibition, depending on the relative contributions of released histamine and somatostatin in each preparation.128,130,131
In the antrum, cholinergic neurons activated by anticipation of the meal stimulate gastrin secretion directly as well as indirectly by suppressing somatostatin secretion (see Fig. 49-10).30,31,34,132–144 In physiologic concentrations, gastrin stimulates parietal cells directly and, more importantly, indirectly by enhancing the secretion of histamine.145,146
As the meal enters the stomach, the same cholinergic neurons are further activated mechanically by high distention and chemically by protein components of the food.141,142,147,148 Initially, the ingested meal acts as a buffer of secreted acid. The decrease in acidity (i.e., increase in pH) further inhibits somatostatin secretion and thus increases gastrin secretion. Luminal protein activates GRP neurons that directly stimulate gastrin secretion (see Fig. 49-10).79,142 It should be noted that suppression of somatostatin secretion permits an optimal gastrin response.
As the meal empties the stomach, a number of paracrine and neural pathways are activated to restore the inhibitory influence of somatostatin in the fundus/body and antrum and hence restrain acid secretion (see Fig. 49-10). First, a stimulatory paracrine pathway linking gastrin to antral somatostatin cells is activated that acts to restore antral somatostatin secretion after release of gastrin.67 Second, there is less activation of cholinergic neurons by anticipation of the meal as well as by protein and distention. Third, as distention decreases, VIP neurons are preferentially activated that stimulate somatostatin secretion.141 Fourth, as the buffering capacity of the meal is lost, antral and fundic somatostatin cells (via sensory CGRP neurons) are exposed to the full stimulatory effect of luminal acid. Fifth, enterogastrones released from the small intestine, such as CCK, stimulate somatostatin secretion. The resultant increase in antral and fundic somatostatin secretion attenuates gastrin and acid secretion and restores the basal interdigestive state. This state is marked by the continuous restraint exerted on G (gastrin), ECL (histamine), and parietal (acid) cells by contiguous somatostatin cells (see Figs. 49-9 and 49-10). A decrease in this restraint is sufficient to again initiate acid secretion.
HELICOBACTER PYLORI–INDUCED PERTURBATIONS IN ACID SECRETION (see also Chapter 50)
Acute infection with H. pylori (HP) results in hypochlorhydria,149–152 whereas chronic infection results in either hypo- or hyperchlorhydria (Fig. 49-16). Appreciation of the pathways discussed earlier provides some insight into the mechanisms whereby HP colonizes the stomach and may lead to ulceration.
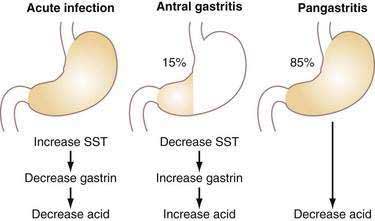
The decrease in acid secretion during acute HP infection is thought to facilitate survival of the organism and its colonization of the stomach.153 The mechanism whereby HP inhibits acid secretion is multifactorial and includes (1) direct inhibition of the parietal cell (and perhaps ECL cell) by a constituent of the bacterium (e.g., vacuolating cytotoxin, lipopolysaccharide, or acid-inhibitory factor) and (2) indirect inhibition of parietal cell function as a result of changes in cytokines as well as hormonal, paracrine, and neural regulatory mechanisms.154–157 HP itself inhibits human H+,K+-ATPase α-subunit gene expression.158
Related posts:
 Surgical Peritonitis and Other Diseases of the Peritoneum, Mesentery, Omentum, and Diaphragm
Surgical Peritonitis and Other Diseases of the Peritoneum, Mesentery, Omentum, and Diaphragm
 Hepatitis Caused by Other Viruses
Hepatitis Caused by Other Viruses
 Hemochromatosis
Hemochromatosis
 Gastrointestinal and Hepatic Complications of Solid Organ and Hematopoietic Cell Transplantation
Gastrointestinal and Hepatic Complications of Solid Organ and Hematopoietic Cell Transplantation
 Nutritional Assessment and Management of the Malnourished Patient
Nutritional Assessment and Management of the Malnourished Patient
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


