Chapter 1A Embryologic development of the liver, biliary tract, and pancreas
Overview of Liver and Biliary Tract Development
Developmental biologists have often marveled at the enormous regenerative capacity of the liver after injury, and such growth represents one of the fastest by mammalian tissues. Presumably this process is based on the recapitulation of embryonic signals in the liver, but our understanding of the mechanisms of liver development is relatively poor. The liver is the largest internal organ and consists of diverse cell types that arise from various embryologic origins. The two principle cell types of the liver are the hepatocytes, which comprise nearly 70% of the mass of the adult organ, and the cholangiocytes. Both cells are derived from the embryonic endoderm. Other cell types of the liver include hematopoietic, Kuppfer, stromal, and stellate cells, all of which are of mesodermal origin (Fig. 1A.1). Despite their homogeneous appearance, hepatocytes do not all function identically; they perform various roles, depending on their physical location within a hepatic lobule, the primary functional unit of the liver. Thus, liver development entails not only hepatocyte and cholangiocyte differentiation but also further cellular differentiation within the hepatocyte population (Fig. 1A.1).
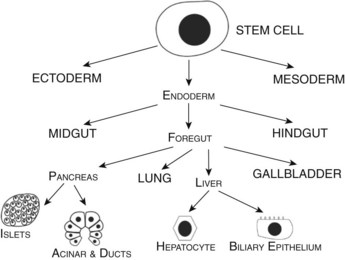
FIGURE 1A.1 Cell lineage schematic for hepatic and pancreatic development from a multipotent progenitor stem cell.
During the third week of gestation, the liver primordium first appears as an outgrowth of the ventral foregut endoderm at the caudal end of the foregut. The proliferation of the epithelial cells in this liver bud leads to its growth and branching with resulting expansion of the growing bud into the surrounding mesenchyme, giving rise to the liver and intrahepatic biliary tree. As it grows caudally, traversing the septum transversum, the persistent connection between the branching epithelium and the foregut develops into the extrahepatic bile ducts and gallbladder (Sadler & Langman, 2006). The hepatoblasts eventually differentiate into hepatocytes and cholangiocytes. The final liver structure continues to develop through the postnatal period. In addition to giving rise to the liver and biliary tract, the proximal endoderm (foregut) also gives rise to respiratory epithelium and the glandular cells of the thyroid, thymus, and pancreas.
Endodermal Patterning
Embryologically, the liver, biliary tract, and pancreas develop through a series of reciprocal interactions between the endoderm and the surrounding mesenchyme. The primitive gut tube, which is derived from the endodermal germ layer during gastrulation, is divided into the foregut, midgut, and hindgut domains, each of which gives rise to specialized regions (Grapin-Botton, 2005). This specialization is initiated by transcription factors expressed in different regions, such as pancreatic and duodenal homeobox gene 1 (PDX1) and pancreas-specific transcription factor 1a (PTF1A), whose coexpression in the endoderm gives rise to the pancreas (Fig. 1A.2) (Grapin-Botton, 2005; Moore-Scott et al, 2007). The definitive endoderm is formed at the ventral side of the vertebrate embryo during gastrulation. Evagination of the endoderm at the anterior end of the embryo generates the ventral foregut, which will eventually give rise to the liver, lung, thyroid, and the ventral pancreas. The dorsal region of the definitive endoderm develops into the intestines and the dorsal pancreas (see Fig. 1A.2).
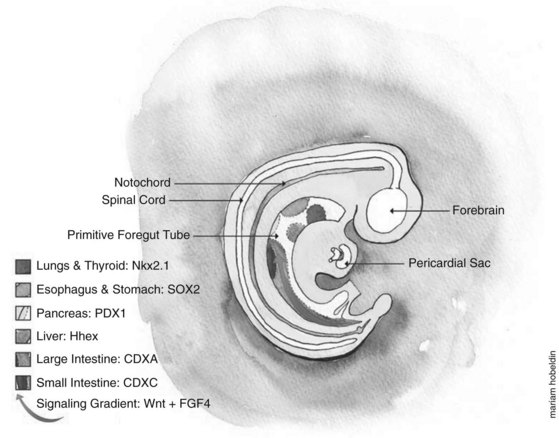
This complex dialogue between the endoderm and mesoderm appears to be critical for the final patterning of the gut tube, where several signaling pathways have been implicated in the regulation of foregut endoderm development, promoting specification along its anterior–posterior axis for organs such as the thyroid, lung, liver, and pancreas. The plasticity of the endoderm was demonstrated by experimentally recombining the posterior mesoderm with early foregut endoderm, leading to repression of liver and pancreatic development in favor of intestinal development (Wells & Melton, 2000; Kumar et al, 2003).
Specific signals from the surrounding mesoderm in the foregut region lead to hepatic specification and subsequent morphogenesis (Guadi et al, 1996). The molecular pathway linking endodermal patterning to the initiation of liver and pancreatic development has been partially elucidated by recent studies in the Xenopus model, supporting a role for FGF4 and WNT signaling from the posterior mesoderm in inhibiting foregut fate and promoting hindgut formation; these signals are inhibited in the anterior endoderm to allow foregut development (McLin et al, 2007). β-Catenin repression in the anterior endoderm is specifically necessary to initiate liver and pancreas development; β-catenin repression in the endoderm then initiates reciprocal signaling with the mesoderm, leading to hepatic induction. To illustrate β-catenin’s role in this process, forced β-catenin expression in the anterior endoderm led to downregulation of the hematopoietically expressed homeobox gene (HHEX) and inhibition of liver formation, whereas repressing β-catenin in the posterior endoderm—future hindgut that normally expresses β-catenin—induced ectopic HHEX expression and ectopic liver bud initiation (McLin et al, 2007). HHEX is a direct target of β-catenin and is one of the earliest foregut markers (Thomas et al, 1998) that is essential for ectopic and normal liver and ventral pancreas development in mice (Keng et al, 2000; Bort et al, 2004). In addition to repressing HHEX, β-catenin also downregulates other foregut markers for liver (for1) (Seo et al, 2002), pancreas (PDX1), lung/thyroid (NKX2-1) (Goss et al, 2009), and intestine (Endocut) (Costa et al, 2003). Similarly, inhibiting β-catenin in the posterior mesoderm led to ectopic expression of other liver and pancreas markers (for1, PDX1, elastase, and amylase) along with inhibition of the intestinal marker Endocut (McLin et al, 2007).
Studies in mouse and chick models demonstrated the need for an FGF2 signal from the cardiogenic mesoderm and bone morphogenetic proteins (BMPs) from the septum transversum mesenchyme (Rossi et al, 2001; Zhang et al, 2004) for liver development. In addition, WNT2B has also been shown to be an important signal for liver specification in Zebrafish (Ober et al, 2006).
Other examples of transcription factors implicated in foregut patterning include the HOX genes. HOXD13 is expressed in the hindgut of mouse and chick embryos (Yokouchi et al, 1995; Roberts et al, 1995; Dolle et al, 1991). Knockout mice homozygous and null mutant for HOXD13 develop with hindgut defects (Kondo et al, 1996). Similarly, when HOXD13 is misexpressed in chick embryo midgut mesoderm, hindgut features are seen in the midgut epithelium (Roberts et al, 1998). The exact mechanism of how the HOX genes induce endodermal patterning remains unknown, as the exact downstream targets are not clear. In addition to HOX genes, retinoic acid signaling has been implicated in this foregut patterning, probably through regulating HOX gene expression. Specifically, retinoic acid signaling appears to specify the position of the pancreatic domain in the foregut of Xenopus, Zebrafish, and mice (Huang et al, 1998; Chen et al, 2004; Martin et al, 2005; Stafford et al, 2004). Goss and colleagues (Goss et al, 2009) demonstrated that WNT2 and WNT2B play an essential role in specifying lung endoderm without affecting the specification of other foregut-derived tissues. WNT2/2B double-knockout (DKO) mutants display complete lung agenesis. NKX2-1, the earliest known transcription factor marker for the developing lung, was also lost from the anterior foregut, confirming the loss of trachea and lung development.
Hepatic Competence
Hepatic competence, or the ability to form liver from the foregut endoderm, is considered the first step in a two-step process for the specification of the liver in vertebrates. Competence is a prerequisite for the endoderm to respond to specific signals, such as Fgfs from the surrounding mesoderm, which then leads to the second step, the induction of liver specific genes such as ALB (albumin). This “competence” is facilitated through the FOXA gene transcription factor family that includes FOXA1 and FOXA2 (forkhead box proteins A1 and A2). FOXA gene expression precedes the induction of the hepatic program by FGF signals in the endoderm, and FOXA2 binding revises α-fetoprotein (AFP) gene transcription in vitro (Crowe et al, 1999). Deletion of FOXA1 and FOXA2 led to no liver bud formation with the loss of AFP expression in the ventral foregut, which would indicate that hepatic specification had failed to initiate (Lee et al, 2005). The forkhead box (FOX) proteins are an extensive family of transcription factors that share homology with the winged helix/forkhead DNA binding domains (Kaestner et al, 2000) that play important roles in regulating expression of genes involved in cellular differentiation, proliferation, transformation, and metabolic homeostasis (Wang et al, 2001; Zaret et al, 1999; Duncan, 2000). In vitro explant cultures demonstrated that Fgf was sufficient to induce ventral foregut endodermal cells to differentiate into hepatoblasts (Guadi et al, 1996). However, when FOXA1/FOXA2-deficient endoderm was cultured with exogenous Fgf2, no liver expression was seen (Lee et al, 2005). This result would indicate the need for FOXA1 and FOXA2 for the onset of hepatogenesis. In vivo DNA-binding studies revealed that the liver-specific albumin gene ALB has an important upstream regulatory binding site for FOXA factors (Guadi et al, 1996; Bossard & Zaret, 1998). Prior to FOXA or GATA4 binding, the ALB gene is transcriptionally silent with a closed chromatin. After binding with FOXA and GATA4, the chromatin domain is thought to become exposed (Cirillo et al, 2002), thus increasing the ability of the gene to be activated. By the E9.5 gestation stage, other transcription factors—CCAAT/enhancer binding protein-β (CEBPB) and NF1, nuclear factor 1—bind to sites adjacent to the FOXA site, and as a result, the albumin gene becomes active (Zaret, 2002). The critical binding of FOXA to chromatin increases the ability of the gene expression to be activated by allowing other transcription factors to bind, hence the hepatic competence.
Hepatic Induction
Fgf from the cardiogenic mesoderm and Bmps from the septum transversum mesenchyme have been implicated in the induction of liver fate in mouse and chick embryonic endoderm. In vitro studies were performed in which the ventral foregut and cardiogenic mesoderm were cocultured in the presence of fibroblast growth factor inhibitors, resulting in the inhibition of liver induction (Jung et al, 1999). Culturing foregut endoderm without the cardiogenic mesoderm in the presence of a low concentration of exogenous FGF2 (2 to 5 ng/mL) rescued hepatic gene expression (Jung et al, 1999) and simultaneously suppressed the expression of the pancreas program (PDX1 expression) (Deutsch et al, 2001), whereas a high concentration of FGF2 (10 to 500 ng/mL) induced NKX2-1, an early marker for respiratory epithelium but not albumin gene expression (Serls et al, 2005). Ventral foregut endoderm cocultured with noggin (Nog), a Bmp inhibitor, inhibited hepatic gene induction, an effect that was rescued when Bmp2 or Bmp4 were added. Despite these results, however, embryos that were homozygous mutant for BMP4 still exhibited normal hepatic gene induction (Rossi et al, 2001; Smith & Harland, 1992). These data indicate that the cardiac mesoderm, a source for FGF signaling, is crucial for the hepatic induction from the ventral foregut endoderm and subsequent morphogenesis, whereas Bmp aids in the process as well (Fig. 1A.3).
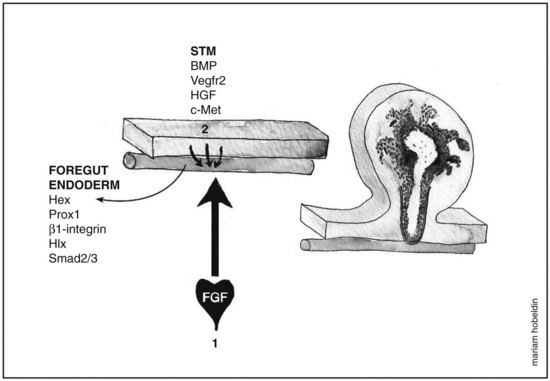
Morphogenesis of the Hepatic Bud
Following hepatic specification (E8.5 to E9.0), the liver starts to express liver-specific genes (ALB, AFP, HNF4A) that eventually form the liver bud. Hepatic bud morphogenesis is facilitated by two transcription factors, HHEX (hematopoietically expressed homeobox, discussed above) (Crompton et al, 1992) and PROX1 (prospero-related homeobox 1) (Oliver et al, 1993). HHEX is expressed in the anterior endoderm at E7.0, eventually giving rise to the ventral pancreas and liver. HHEX-null embryos grow without a liver, thyroid and develop forebrain defects at E11.5; however, evidence of endodermal epithelial thickening suggest a possible defect in differentiation as evidence of possible hepatic induction (Martinez-Barbera et al, 2000).
In a separate study on HHEX-null embryos, liver genes ALB and PROX1 were expressed in the ventral foregut endoderm at around E8.5, with the thickening of the hepatic endoderm region at E9.0 being smaller compared with heterozygote embryos. In addition, a significantly lower proliferation rate was seen in the prospective hepatic domain compared with controls, as demonstrated by bromodeoxyuridine (BrdU) staining, with no evidence of apoptosis (Bort et al, 2004). The basal membrane layer, which is rich in laminin, surrounds the hepatic endoderm and degrades around E9.0 to E9.5, so that the hepatocytes start migrating into the septum transversum mesenchyme (STM) to form the liver bud. This degradation is facilitated by the hepatoblasts, which under normal conditions downregulate E-cadherin. However, in PROX1-null mutant embryos, the progenitor liver cells fail to migrate into the STM as a result of excess E-cadherin and basement membrane proteins laminin and collagen 4. The basal lamina fails to degrade, and the cells remain trapped in the hepatic diverticulum with an overall reduction in liver size (Sosa-Pineda et al, 2000). The bulk of the liver lobe lacks hepatocytes, suggesting that the mesenchymal component contributes most of the liver mass in PROX1-null mutant embryos. A similar phenotype is seen with ONECUT1 and ONECUT2 double mutants, which are required for basal lamina degradation (Margagliotti et al, 2007). Furthermore, pharmacologic inhibition of matrix metalloproteinases (MMPs), extracellular matrix remodeling enzymes usually expressed by hepatoblasts and STM cells, inhibits hepatoblast migration in culture (Margagliotti, 2008). To further illustrate the importance of the extracellular matrix in hepatic bud morphogenesis, hepatoblasts deficient in laminin receptor β1-integrin fail to colonize the liver bud (Fassler & Meyer, 1995). These β1-integrins are among those that act as receptors for extracellular matrix proteins, such as laminins and collagens (Hynes, 1992).
The role of endothelial cells in liver organogenesis has also been studied, with the liver vasculature providing a vital source for hematopoiesis in early life. Endothelial cells are positioned as a loose chain of cells interceding between the thickening hepatic epithelium and the STM (Matsumoto et al, 2001). Embryos that were null mutant for VEGFR2 (also known as KDR) had failure of delamination and subsequent migration by the hepatoblasts (Matsumoto et al, 2001). These results imply that the endothelial cells interact with nascent hepatic cells and aid in liver bud outgrowth.
Liver Bud Growth
The STM cells are closely related to the ventral endoderm and contribute to hepatic induction and growth. This epithelial–mesenchymal interaction is essential for the formation of the liver bud as well as for further expansion and differentiation. During this phase (E9.5 to E15), the liver bud undergoes a tremendous amount of growth and also becomes an important site for hematopoiesis. Factors and signals regulating this critical stage arise from both the hepatic mesenchyme and the STM. These signals include Fgf and Bmp, which promote liver growth in addition to their role in hepatic specification. Bmp4 is strongly expressed in the STM and continues to be expressed at E9.0, during which the liver bud migrates into the STM (Rossi et al, 2001). BMP4-null mutant embryos were developed to assess for a role in liver organogenesis. These null-mutant mice had a delay in the growth of the liver bud, which would indicate that Bmp constitutes an important growth signal for the liver (Rossi et al, 2001).
Another factor that has been implicated in liver growth is HLX, which is expressed prominently in the visceral mesenchyme (STM), into which the liver will expand (Hentsch et al, 1996). HLX-null embryos exhibited severe liver hypoplasia without affecting liver specification (Hentsch et al, 1996). A similar finding was observed in embryos that were null mutant for hepatocyte growth factor (Hgf) (Schmidt et al, 1995). In contrast to HLX−/− embryos, apoptosis was the underlying cause for the severe reduction in liver size. Hgf is produced by the cells lining the sinusoids, which are of mesenchymal origin, mediating its effects through the c-Met tyrosine kinase receptor produced by hepatocytes (Schmidt, 1995). TGF-β signaling is also involved in liver outgrowth. It mediates its signals through SMAD2 and SMAD3, which after phosphorylation translocate to the nucleus to either upregulate or downregulate gene expression. Embryos that were heterozygous mutants for both SMAD2 and SMAD3 exhibited severe liver hypoplasia as a result of a decrease in β1-integrin expression (Weinstein et al, 2001). Intriguingly, this phenotype was rescued when HGF, a potent hepatotrophic growth factor, was added to the culture medium. Presumably, this was a result of the restoration of β1-integrin expression (Kagami et al, 1996), which plays an important role in hepatocytic adhesion to the extracellular matrix; TGF-β and HGF are known to induce the gene (Kagami et al, 1996; Kawakami-Kimura et al, 1997).
Overview of Hepatoblast Differentiation
Hepatoblasts begin to differentiate into mature hepatocytes and biliary epithelial cells at around E13.5. Prior to differentiation, the hepatoblasts express genes for adult hepatocytes (ALB, HNF4A), biliary epithelial cells (KRT19), and fetal liver (AFP) (Lemaigre, 2003; Shiojiri et al, 1991). Hepatoblasts in proximity to the portal vein form a bilayer and eventually differentiate into biliary epithelial cells, upregulating biliary-specific cytokeratin-19 (KRT19) and downregulating the other hepatic genes in the process. This bilayer around the portal vein begins to form focal dilations incorporated into the portal mesenchyme to form intrahepatic biliary ducts at E17.0 until birth. Those parts of the ductal bilayer plate not involved in the formation of the ducts progressively regress, and the rest of the hepatoblasts differentiate into mature hepatocytes, arranged in hepatic cords with the bile canaliculi on the apical surfaces (Lemaigre et al, 2003). The bile is produced by the hepatocytes and is secreted into the canaliculi, which are connected to the network of intrahepatic biliary ducts. The bile then flows to the hepatic ducts, transits through the cystic duct, and is stored in the gallbladder; eventually, the bile is excreted into the bowel via the common bile duct. The biliary epithelial cells delineate the lumen of the intrahepatic and extrahepatic biliary tree (hepatic, cystic, and common bile ducts) and the gallbladder (Fig. 1A.4).
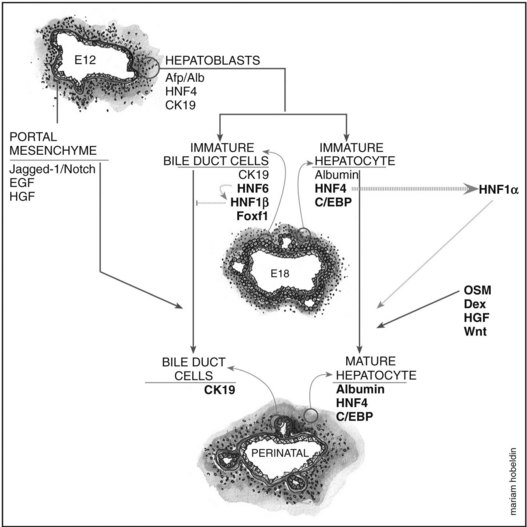
Biliary Epithelial Cell Differentiation and Formation of the Ductal Plate
The exact origin of the biliary epithelial cells has been greatly debated; however, the popular school of thought is that they are derived from bipotential hepatoblasts that can differentiate into either hepatocytes or biliary epithelial cells. This theory is based on the observation that immature hepatoblasts coexpress markers of both hepatocytes (ALB) and biliary epithelial cells (KRT19). The biliary-specific marker KRT19 becomes strongly expressed at a later gestational age, as the cells become ductal cells, whereas other cells transiently express the hepatocyte markers ALB and AFP as they develop into mature hepatocytes (Shiojiri et al, 1991). This theory was further supported when embryonic liver was transplanted into the testes of syngeneic animals, before the emergence of intrahepatic biliary ducts, as it subsequently gave rise to both hepatocytes and typical bile ducts (Shiojiri et al, 1991). However, this finding does not exclude the possibility that ductal cells may be derived from other embryonic cell types. Suzuki and colleagues (Suzuki et al, 2002) used in vitro studies on hepatic “stem” cells from E13 embryonic livers identified as having self-renewing capability and multilineage differentiation potential, to show that these cells could form differentiated hepatocytes, biliary epithelial cells, pancreatic, and intestinal cells.
The first step in triggering the initiation of the transition from a hepatoblast to a biliary epithelial cell is thought to be facilitated through the ONECUT transcription factor hepatocyte nuclear factor 6 (HNF6), which is expressed in the biliary epithelial cells of the developing intrahepatic biliary ducts and in hepatoblasts, gallbladder primordium, and the extrahepatic bile ducts (Landry et al, 1997; Rausa et al, 1997). HNF6−/− embryos displayed severe biliary anomalies that resulted in an absent gallbladder and the extrahepatic bile duct network being replaced with an enlarged structure connecting the liver to the duodenum. In the intrahepatic biliary ducts, however, it caused abnormal differentiation of biliary epithelial cells, resulting in cholestasis. Closer histologic examination revealed an increased number of KRT19-positive cells compared with controls at E13.5, with the development of abnormal large cysts at E15.5 to E16.5 that contained an epithelium of KRT19-expressing cells. These abnormal cysts are similar to those seen in Caroli disease, a disorder with ductal plate malformation and ectasias. Although HNF6 might play a role in the disease, not all of the malformations seen in HNF6−/− mice are manifested in Caroli disease, such as the absence of the gallbladder. The abnormal increase in KRT19-positive cells at E13.5 lacked any proliferative marker, suggesting that they are postmitotic, resulting from an increased number of hepatoblasts that have differentiated toward a biliary lineage prematurely (Clotman et al, 2002). In addition, the excess KRT19-positive biliary epithelial cells formed cordlike extensions within the liver parenchyma, rather than being restricted to the vicinity of the portal vein, as seen in the controls. This observation supports the role of HNF6 in controlling the differentiation of hepatoblasts into biliary epithelial cells and the morphogenesis of the intrahepatic biliary ducts, confining biliary epithelial cells to the periportal area. A similar morphologic defect of intrahepatic biliary ducts was observed in HNF1B−/− embryos. These findings suggest that HNF6 controls intrahepatic biliary duct development via HNF1B (Clotman et al, 2002).
With regard to mesenchymal–epithelial induction of liver primordium and gallbladder, studies have suggested that the mesenchyme contributes to biliary epithelial cell differentiation, where differentiation of hepatoblasts into biliary epithelial cells was stimulated when cocultured with hepatic or lung mesenchyme (Shiojiri & Koike, 1997). It was also noted in the HNF6−/− study that biliary epithelial cell differentiation occurred at the interface between the portal mesenchyme and the liver parenchyma (Clotman et al, 2002). A recent study revealed that the forkhead box f1 (FOXF1) transcription factor may play an important role in mesenchymal–epithelial signaling, an interface required for the development of organs derived from foregut endoderm, such as the pancreas, liver, gallbladder, and lung (Kalinichenko et al, 2002). FOXF1 expression is restricted to the gallbladder mesenchyme and STM. In FOXF1+/−, the gallbladder develops severe structural abnormalities with significant reduction in size and reduced mesenchymal cell numbers, an absent biliary epithelial cell layer, and a deficient external smooth muscle layer. The reduction in mesenchymal cell numbers was attributed to the reduction in the vascular cell-adhesion molecule (VCAM1) and cell-adhesion α5 integrin (ITGA5) genes, both of which are essential for mesodermal formation (Mahlapuu et al, 2001; Yang et al, 1993). Defective smooth muscle layer formation was attributed to diminished levels of platelet-derived growth factor receptor α (PDGFRA), which is essential for smooth muscle cell differentiation (Jacob et al, 1994). FOXF1 is not expressed in intrahepatic biliary duct mesenchyme in wild-type mice, and no defects were seen in the intrahepatic biliary ducts of FOXF1+/− mice; however, FOXF1 mRNA levels in the liver were increased, suggesting that it may be a compensatory mechanism to prevent defects in the liver (Kalinichenko et al, 2002). All these data suggest that FOXF1 is crucial for the development of the extrahepatic bile ducts and gallbladder, with the mesenchyme playing an essential role in biliary epithelial cell differentiation. The interaction between the biliary epithelial cells and the extracellular matrix is also thought to contribute to biliary epithelial cell differentiation.
Integrins are membrane receptors for extracellular matrix proteins that play an important role in mediating the interaction between differentiating cells and the extracellular matrix (Hynes, 1992; Couvelard et al, 1998). Hepatoblasts express integrin heterodimers containing the β1 subunit (α1β1, α5β1, α6β1, and α9β1), and when hepatoblasts differentiate into immature intrahepatic biliary epithelial cells when in contact with the mesenchyme, the morphology of the integrins changes. The primitive intrahepatic biliary epithelial cells upregulate α6β1 expression and lose α1β1, while acquiring several other integrin dimers not previously expressed on hepatoblasts, including α2β1, α3β1, α5β1 and α6β4 (Couvelard et al, 1998). This change is likely due to the different pericellular environment. Intrahepatic biliary epithelial cells are contacted by a basement membrane containing collagen, entactin, and laminin (Desmet, 1985); however, hepatocytes are surrounded by the perisinusoidal matrix, which is devoid of laminin and entactin (Schuppan, 1990). The increase in α6β1 expression—along with the acquisition of biliary-specific expression of α2β1, α3β1, and α6β4, which are integrin receptors for laminin—correlated with the deposition of laminin at the contact points of the portal mesenchyme with the ductal plate (Couvelard et al, 1998).
Remodeling of the Ductal Plate
As mentioned earlier, the double-layered ductal plate around the portal vein begins to form focal dilations incorporated into the portal mesenchyme to form intrahepatic biliary ducts, while the parts of the ductal bilayer plate not involved in the formation of the ducts progressively regresses. This mechanism of regression is thought to be carried out by apoptosis (Terada & Nakanuma, 1995; Sergi et al, 2000). Cell-matrix interactions are also thought to contribute to the remodeling process. Tenascin was found to be expressed in the mesenchyme around the biliary epithelial cells of primitive ducts migrating into the mesenchyme. In contrast, tenascin was absent in the mesenchyme of peripheral ducts (Terada & Nakanuma, 1994).
Tubulogenesis of ductal cells is thought to be controlled by soluble factors secreted from hepatocytes or biliary epithelial cells. When coculturing human biliary epithelial cells with hepatocytes, a marked ductular morphogenic response was induced, and the biliary epithelial cells formed well-organized luminal ducts. This result was reproduced when biliary epithelial cells were grown in a conditioned medium from previous hepatocyte and biliary epithelial cell coculturing (Auth et al, 2001); however, it remains to be seen whether soluble factors do contribute to tubulogenesis in in vivo studies (Fig. 1A.5).
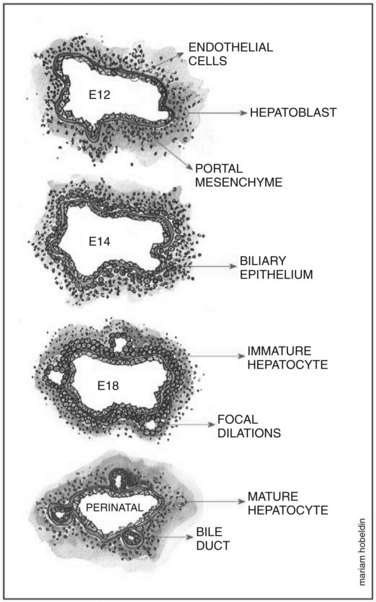
Developmental Relationship Between the Ducts, Vessels, and Mesenchyme of the Portal Tract
A functional relationship appears to exist between the contents of the portal tract (bile duct, hepatic artery, portal vein) based on observations of a number of human diseases termed ductal plate malformations. These diseases include biliary atresia, Caroli disease, and Meckel and Alagille syndromes, in which abnormal biliary ducts are associated with anomalies of the portal mesenchyme and of the portal blood vessels (Lemaigre, 2003). This association was also demonstrated in studies on Notch pathway defects. In Alagille syndrome, an autosomal dominant disease, bile ducts are absent in the portal tract associated with an increased number of arteries and fibrosis. Haploinsufficiency of JAG1, a Notch receptor ligand, is associated with Alagille syndrome, where JAG1 is persistently expressed in the ductal epithelium in humans (Li et al, 1997; Louis et al, 1999). It is also expressed in the endothelial cells of the developing portal vasculature (Crosnier et al, 2000). The animal model for Alagille syndrome was replicated in double-heterozygous mice for mutations in the JAG1 and NOTCH2
Related posts:
 Preoperative and postoperative nutrition in hepatobiliary surgery
Medical management of bleeding varices: Primary and secondary prophylaxis
Preoperative and postoperative nutrition in hepatobiliary surgery
Medical management of bleeding varices: Primary and secondary prophylaxis
 Nonhepatic surgery in the cirrhotic patient
Nonhepatic surgery in the cirrhotic patient
 Liver transplantation for cholangiocarcinoma and other neoplastic diseases
Liver transplantation for cholangiocarcinoma and other neoplastic diseases
 Distal splenorenal shunt
Distal splenorenal shunt
 Interventional endoscopy: Technical aspects
Interventional endoscopy: Technical aspects
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


