Disorders of Sex Development: Introduction
What defines our sexuality is a complex interaction between our genetic makeup, environmental stimulus, and cultural influences. The origins of our sexuality occur at the time of conception when the genetic material from two sources of the opposite sex coalesces into a new individual. From that moment, sexual differentiation occurs by a highly organized process. Sex chromosomes and autosomes dictate the development of gonads; the gonads in turn produce hormones, which then direct the development of the internal and external genitalia. Disorders of sex development (DSD) or differentiation arise from abnormalities in chromosomes, gonadal development, or hormonal production/activity.
Patients with DSD become apparent (1) during the newborn period as having ambiguous genitalia or a discordant phenotypic from the genotype found at the time of amniocentesis, (2) as having inappropriate pubertal development, (3) as having delayed pubertal development, or (4) later in life as having infertility.
Normal Sexual Differentiation
The genetic material necessary for the development of the male phenotype is normally located on the short arm of the Y chromosome (Wilson et al, 1981). The critical gene or sex-determining region on the Y chromosome is known as the SRY region. The gene products of the SRY genetic cascade direct the development of the testis by interacting with multiple other genes such as SOX-9 (Conte and Grumbach, 2007). Genetic information that is known to be necessary for male and female development beyond gonadal differentiation is located on the X chromosome and on the autosomes.
The gonads develop from the urogenital ridges (Figure 43–1), which are formed during the 4th week of gestation by the proliferation of the coelomic epithelium and condensation of the underlying mesenchyme along the mesonephros. The germ cells, located in the endoderm of the yolk sac, migrate to the genital ridges. At the early stage of development, the gonad is bipotential, capable of forming into either a testis or an ovary. During the 6th–7th week of gestation, at least four different genes, Wilms’ tumor suppressor gene (WT-1), Fushi-Tarza Factor-1 (FTZ-F1), steroidogenic Factor-1 (SF-1), and LIM-1, induce the development of the testis. The primordial germ cells differentiate into the Sertoli cells and associated Leydig cells, which aggregate into spermatogenic cords. Loose mesenchymal tissue condenses into a thick layer, the tunica albuginea, which surrounds the testis and separates its connection with the coelomic epithelium, thereby preventing further migration of mesonephric cells into the testis.
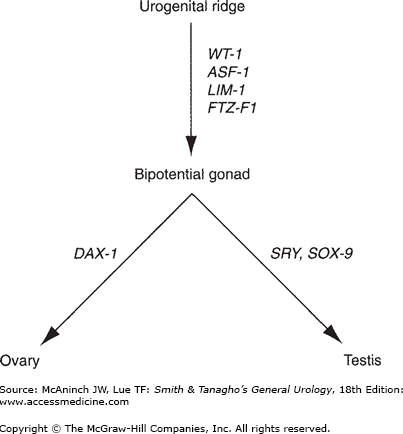
Classic teaching is that the female phenotype is the default developmental pathway in the absence of the SRY cascade. It is now known that at least one gene, dosage-sensitive sex reversal (DAX-1), is essential for ovarian development. DAX-1 is located on the short arm of the X chromosome. The gene products of SRY and DAX-1 compete to stimulate the steroidogenic acute regulatory protein (StAR). The StAR protein is the first step in steroidogenesis, facilitating the conversion of cholesterol to pregnenolone. In the normal XY male, SRY overwhelms the one functional DAX-1 gene, stimulating testicular development and subsequent testosterone production. In the normal XX female, two DAX-1 genes are present without the competitive SRY, downregulating StAR, hence inhibiting testicular development, which results in ovarian development. In the fetal ovaries, the germ cells differentiate and are arrested in the last phase of meiotic prophase, forming the oocytes. The cells in the genital ridges develop into granulosa cells, which surround the oocytes and complete the formation of the ovaries.
At 3.5 weeks’ gestation, the Wolffian system appears as two longitudinal ducts connecting cranially to the mesonephros and caudally draining into the urogenital sinus (Figure 43–2). At approximately the 6th week of gestation, the Müllerian duct develops as an evagination in the coelomic epithelium just lateral to the Wolffian duct.
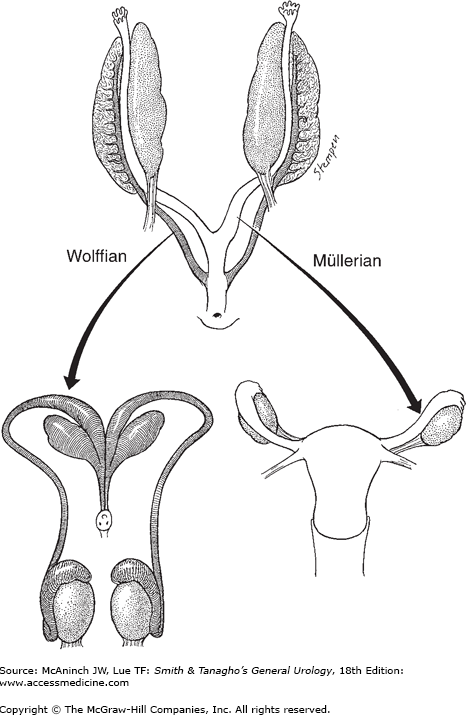
During the 8th–9th week of gestation, Sertoli cells of the fetal testis secrete a glycoprotein, Müllerian-inhibiting substance (MIS), or anti-Müllerian hormone. This protein induces the regression of the Müllerian ducts through the dissolution of the basement membrane and condensation of mesenchymal cells around the Müllerian duct. Because MIS acts locally, Müllerian duct regression occurs only on the ipsilateral side of the fetal testis producing this hormone. MIS also induces the formation of seminiferous tubules and further differentiation of the testis. At the 9th or 10th week of gestation, the Leydig cells appear in the testis and begin to synthesize testosterone. This hormone transforms the Wolffian duct into the male genital tract, which is completed by the end of the 11th week of gestation.
Beginning in the 9th week of gestation, testosterone also induces the development of the external genitalia (Figure 43–3) from the genital tubercle, urogenital sinus, and genital swellings (Jirasek et al, 1968). At the molecular level, testosterone is converted to 5α-dihydrotestosterone (DHT) by the microsomal enzyme, type 2 5α-reductase, for complete differentiation of the penis with a male-type urethra and glans (Wilson et al, 1993). Testosterone dissociates from its carrier proteins in the plasma and enters cells via passive diffusion. Once in the cell, testosterone binds to the androgen receptor (AR) and induces changes in conformation, protecting it from degradation by proteolytic enzymes. This conformational change is also required for AR dimerization, DNA binding, and transcriptional activation, all necessary for testosterone to be expressed. Androgen binding also displaces heat shock proteins, possibly relieving constraints on receptor dimerization or DNA binding. After entering the nucleus, the AR complex then binds androgen response element DNA regulatory sequences within the androgen responsive genes and activates them. DHT also binds the AR, with enhanced androgenic activity, in part because of its slow dissociation rate from the AR.
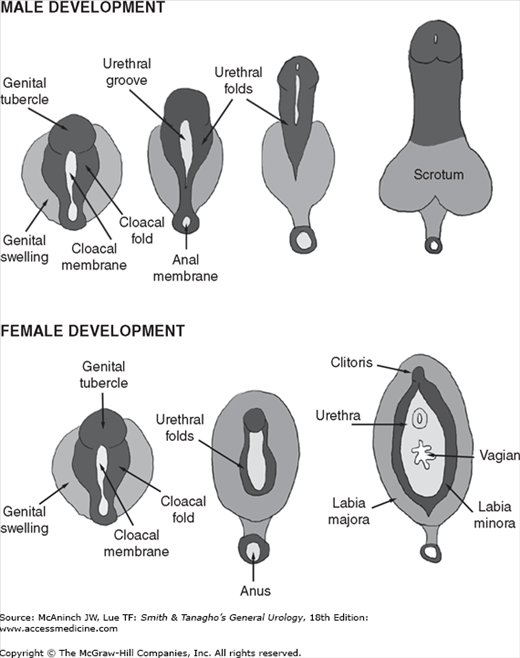
DHT then binds to nuclear receptors, forming a complex that regulates the transformation of these tissues into the glans penis, penile and cavernous urethra, Cowper’s glands, prostate, and scrotum. Between the 28th and 37th week of gestation, testicular descent into the scrotum begins. While the mechanism of this process is not completely understood, it is clearly androgen dependent.
The female internal genitalia develop from the Müllerian ducts. Without the hormones produced by the testis, the Wolffian ducts regress at the 9th week of gestation. At the same time, the Müllerian ducts begin to differentiate; the cranial portions form the fallopian tubes, while the caudal portions fuse to form the uterus, cervix, and the upper portion of the vagina. Concurrently, the external genitalia defined as the lower portion of the vagina, the vestibule, Bartholin and Skene glands, the clitoris, and labia minora and majora develop from the urogenital sinus and genital tubercles. Like the testis, the ovary undergoes a partial transabdominal descent. However, transinguinal descent of the ovary does not occur, leaving the ovaries just below the rim of the true pelvis. The role of estrogen in the differentiation of the female phenotype is unclear.
Formation of the external male genitalia is a complex developmental process involving the SRY genetic programming, cell differentiation, hormonal signaling, enzyme activity, and tissue remodeling. By the end of the 1st month of gestation, the hindgut and future urogenital system reach the ventral surface of the embryo at the cloacal membrane. The urorectal septum divides the cloacal membrane into a posterior, or anal, half and an anterior half, the urogenital membrane. Three protuberances appear around the latter. The most cephalad is the genital tubercle. The other two, the genital swellings, flank the urogenital membrane on each side. Up to this point, the male and female genitalia are essentially indistinguishable. Under the influence of testosterone in response to a surge of luteinizing hormone from the pituitary, masculinization of the external genitalia takes place. One of the first signs of masculinization is an increase in the distance between the anus and the genital structures, followed by elongation of the phallus, formation of the penile urethra from the urethral groove, and development of the prepuce.
At 8 weeks’ gestation, the external genitalia remain in the indifferent stage. The urethral groove on the ventral surface of the phallus is between the paired urethral folds. The penile urethra forms as a result of fusion of the medial edges of the endodermal urethral folds. As development progresses, the ectodermal edges of the urethral groove begin to fuse to form the median raphe (Figure 43–4A). By 11–12 weeks, the coronal sulcus separates the glans from the shaft of the penis. By 16 weeks’ gestation, the urethral folds have completely fused in the midline on the ventrum of the penile shaft (Figure 43–4B). Note the normal ventral penile curvature, or chordee, that occurs during development and resolves by the 20th week (Figure 43–4C).
Figure 43–4.
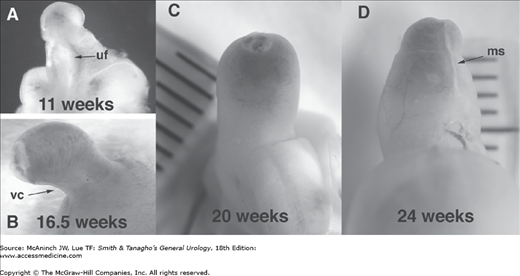
Male human fetal external genitalia during gestation. A: 11 weeks. Note the urethra is open and urethral fold (uf) and groove are prominent in the transillumination view of the phallus. B: 16.5 weeks. Note the normal ventral curvature (vc) as well as the foreskin, which is almost completely formed. C: At 20 weeks’ gestation, penile and urethral development looks complete, with the prepuce covering the glans and the penile curvature resolving. D: At 24 weeks, the prepuce covers the whole glans. Note the midline seam (ms). Note the progression of natural curvature to a straight phallus during development.
The glandular urethra, which consists of a squamous epithelial-lined tube different from the urothelial-lined anterior urethra, also completes its formation during this period. The mechanism of the glandular urethral formation remains controversial. Evidence suggests two possible explanations (Figure 43–5): (1) endodermal cellular differentiation where the glandular urethra forms by an extension of urogenital sinus epithelium that undergoes transdifferentiation versus (Kurzrock et al, 1999) (2) primary intrusion of the ectodermal tissue from the skin of the glans penis. Cross-sectional histologic analysis at 24 weeks’ gestation reveals complete penile development (Figures 43–6A–H). Note the extensive neuronal innervation just above the tunica of the corporeal bodies. Three-dimensional reconstruction of the fetal male penis illustrates the extensive neuronal distribution (Figure 43–7). Note the nerve density in the glans (Figures 43–7E and F).
Figure 43–5.
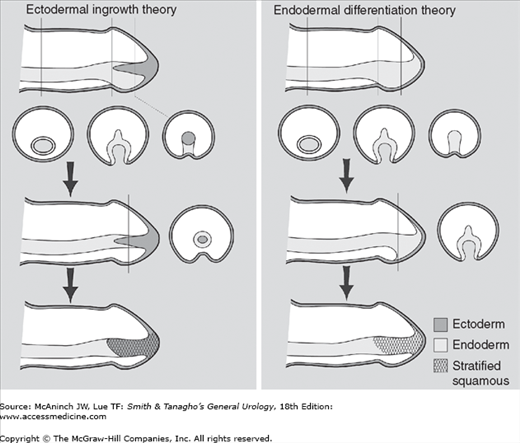
Theories of human penile urethral development. The ectodermal ingrowth theory as described in most textbooks of embryology postulates that the glanular urethra is formed by ingrowth of epidermis. More recent data support the formation of the entire urethra via endodermal differentiation alone.
Figure 43–6.
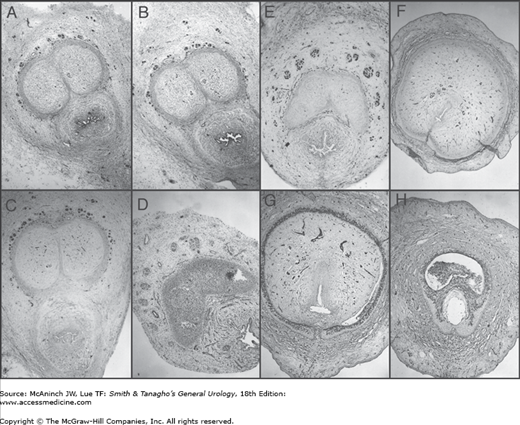
Normal human fetal penis, 24 weeks (A–H). Transverse histologic sections show immunohistochemical localization with the neuronal marker S-100 (25×). Note localization of S-100 nerve marker (dark staining) completely surrounding the cavernous bodies up to the junction with the urethral spongiosum along the penile shaft except at the 12 o’clock position (A–D). On the proximal penis at the point where the corporeal bodies split into two (E) and continue in a lateral fashion inferior and adjacent to the pubic rami, the nerves localize to an imaginary triangular area at the 11 o’clock and 1 o’clock positions. At this point (E), the nerves reach their furthest vertical distance from the corporeal body (approximately one-half the diameter of the corporeal body) and continue (F–G) in a tighter formation at the 11 o’clock and 1 o’clock positions well away from the urethra.
Figure 43–7.
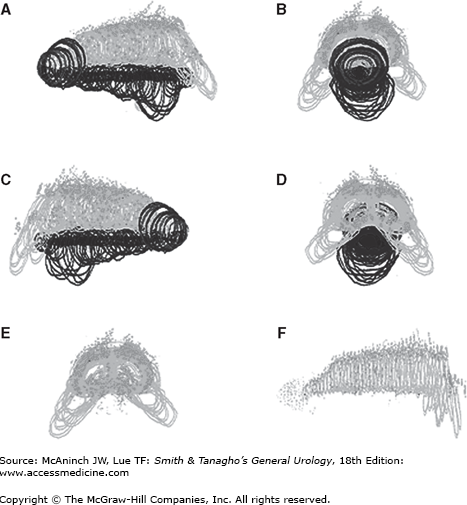
Normal human fetal penis, 45 weeks’ gestation. Four views of a computer-generated three-dimensional reconstruction (A, side; B, front; C, side; D, back, E, front [without urethra]; F, side [without urethra]). Note the nerves along the outside of the tunica of the corporeal bodies and their absence at the 12 o’clock position. Note the impressive glandular innervation in E and F.
Anatomical and immunohistochemical studies advocate the new theory of endodermal differentiation, which shows that epithelium of the entire urethra is of urogenital sinus origin. The entire male urethra, including the glandular urethra, is formed by dorsal growth of the urethral plate into the genital tubercle and ventral growth and fusion of the urethral folds. Under proper mesenchymal induction, urothelium has the ability to differentiate into a stratified squamous phenotype with characteristic keratin staining, thereby explaining the cell type of the glans penis.
The future prepuce is forming at the same time as the urethra and is dependent on normal urethral development. At about 8 weeks’ gestation, low preputial folds appear on both sides of the penile shaft, which join dorsally to form a flat ridge at the proximal edge of the corona. The ridge does not entirely encircle the glans because it is blocked on the ventrum by incomplete development of the glandular urethra. Thus, the preputial fold is transported distally by active growth of the mesenchyme between it and the glandular lamella. The process continues until the preputial fold (foreskin), covers all of the glans, forming a midline seam (Figure 43–4D). The fusion is usually present at birth, but subsequent desquamation of the epithelial fusion allows the prepuce to retract. If the genital folds fail to fuse, the preputial tissues do not form ventrally; consequently, in hypospadias, preputial tissue is absent on the ventrum, and excessive dorsally.
Disorders of abnormal sexual differentiation may be divided into the following three categories.
These result from abnormalities in the number or structure of the sex chromosomes. These abnormalities may arise from nondisjunction, deletion, breakage, rearrangement, or translocation of genetic material on these chromosomes. These disorders are summarized in Table 43–1.
Disorder | Pathology | Chromosomes | Incidence | Gonads | Internal genitalia | External genitalia | Other features | Risk of cancer | Treatment |
|---|---|---|---|---|---|---|---|---|---|
47,XXY (Klinefelter syndrome) | Extra X chromosome | 47,XXY 46,XY/47,XXY | 1 in 500 | Hyalinized testis No spermatogenesis | Wolffian | Male | Gynecomastia Tall stature Mild mental retardation Elevated FSH/LH Low testosterone Elevated estradiol Infertility | Breast Extragonadal germ cell | Supplemental androgens Surgery for severe gynecomastia |
XX male (XX sex reversal) | No Y chromosome Usually TDF (+) | 46,XX | 1 in 20,000–24,000 | Hyalinized testis No spermatogenesis | Wolffian | Male | Gynecomastia Short stature Inc. incidence of hypospadias Normal mental status May be familial | Rare germ cell | Same as Klinefelter |
45,X (Turner’s syndrome) | Absence of X chromosome | 45,X 46,XX/45,X Some contain Y chrom. elements | 1 in 2700 | Streak gonads No germ cells | Müllerian | Immature female | Short stature Little breast development Web neck and other somatic abn. Cardiovascular abn. (ie, coarctation) Renal abn. (Horseshoe or malrotation) Autoimmune dz. (hypothyroid, diabetes) Infertility Amenorrhea | Germ cell Y-chrom mosaic | Supplemental estrogen Removal of streak gonads in Y-chrom. mosaic |
45,X/46,XY DSD (Mixed gonadal dysgenesis) | Incompl. virilization & Müllerian regression | 45,X/46,XY (70%) Undetected mosaic | One testis (usually undescended) and streak gonad | Wolffian and Müllerian | Usually ambiguous 60% reared as female | Somatic features like 45,X | Germ cell | Female —Prophylactic gonadectomy Male —Streak gonads removed —Intra-abd. testis excised unless can be relocated and no ipsilateral Müllerian structure present | |
Ovotesticular DSD (True hermaphrodite) | Unknown | 46,XX (70%) 46,XY (10%) Mosaic | Unknown | Bilateral ovotestis Ovotestis & ovary or testis (40%) One ovary & testis (40%) | Wolffian and Müllerian | Usually ambiguous 70% reared as male | Gynecomastia at puberty Menstruation at puberty May be familial | Rare germ cell | Reconstructive surgery Poss. remove gonads |
These result from abnormalities in gonadal development. In these disorders, the karyotype is normal (ie, 46,XX or 46,XY). However, mutations in the sex chromosomes or autosomes, teratogens, or trauma to the gonads interfere with their normal development. These disorders are summarized in Table 43–2.
Disorder | Pathology | Chromosomes | Incidence | Gonads | Internal genitalia | External genitalia | Other features | Risk of cancer | Treatment |
|---|---|---|---|---|---|---|---|---|---|
Complete gonadal dysgenesis (Swyer Syndrome) | Unknown mutation prevents nl. differentiation of gonads | 46,XX 46,XY | 1 in 8000 | Bilateral streak gonads | Müllerian | Immature female | Normal to tall stature Minimal somatic abn. Female: estrogen def. Male: testosterone def. May be familial | Germ cell in 46,XY | Estrogen supplement Remove gonads in 46,XY |
Absent testes syndrome | Mutation, teratogen or trauma to testis | 46,XY | Unknown | Absent/rudiment testis No streak gonads | Wolffian | Var. virilization | Normal | Usually none | Female —Estrogen supplement —Reconstructive surgery Male —Androgen supplement |
These result from abnormalities in hormonal production or activity. The etiologies include defective synthesis by the gonads, abnormal production by the adrenal glands, the presence of exogenous sources, or abnormalities in receptor activity. These disorders are summarized in Table 43–3.
Disorder | Pathology | Chromosomes | Incidence | Gonads | Internal genitalia | External genitalia | Other features | Urinary steroids | Risk of cancer | Treatment |
|---|---|---|---|---|---|---|---|---|---|---|
46,XX DSD (Female Pseudohermaphrodite) | ||||||||||
3 β-Hydroxysteroid dehydrogenase def. | Excess androgens | 46,XX | Second most common of CAH | Ovary | Müllerian | Mild ambiguous | Severe salt wasting No cortisol No aldosterone | DEAS | None | Replacement mineralocorticoids and glucocorticoids Reconstruction as needed |
11 β-Hydroxy lase def. | Excess androgens | 46,XX | Rare | Ovary | Müllerian | Ambiguous | Hypertension Dec. cortisol Dec. aldosterone | 11 DCS 11 DOC | None | Replacement glucocorticoids |
21 α-Hydroxylase def.—Partial | Excess androgens | 46,XX | 1 in 5000–15,000 | Ovary | Müllerian | Ambiguous | Normal cortisol Inc. aldoster one | 17 OH-P | None | Reconstruction as needed |
—Severe | Excess androgens | 46,XX | Ovary | Müllerian | Ambiguous | Severe salt wasting Dec. cortisol Dec. aldosterone | 17 OH-P | None | Replacement mineralocorticoids and glucocorticoids Reconstruction as needed | |
Excess maternal androgens | Excess androgens | 46,XX | Ovary | Müllerian | Ambiguous | Drugs such as progestational agents Virilizing ovarian Adrenal tumors | None | None | None | |
46,XY DSD (Male Pseudohermaphrodite) | ||||||||||
20,22 Desmolase def. | Defect in testosterone synthesis | 46,XY | Testis | Wolffian | Ambiguous | Severe salt wasting No cortisol No aldosterone | None | None | Replacement mineralocorticoids and glucocorticoids | |
3 β-Hydroxy steroid dehydrogenase def. | Defect in testosterone synthesis | 46,XY | Second most common in CAH | Testis | Wolffian | Ambiguous | Severe salt wasting No cortisol No aldosterone | DEAS | None | Replacement mineralocorticoids and glucocorticoids Reconstruction as needed |
17 α-Hydroxylase def. | Defect in testosterone synthesis | 46,XY | Testis | Wolffian | Ambiguous | Hypokalemic alkalosis Hypertension Dec. cortisol Dec. aldosterone Gynecomastia | CS 11 DCS | None | Replacement glucocorticoids | |
17,20 Desmolase def. | Defect in testosterone synthesis | 46,XY | Rare | Testis | Wolffian | Ambiguous | Normal cortisol and aldosterone | None | Supplemental testosterone | |
17 β-Hydroxysteroid dehydrogenase def. | Defect in testosterone synthesis | 46,XY | Most common | Testis | Wolffian | Ambiguous | Virilization and puberty | ASD | None | Decision reared as female or male |
5 α-Reductase def. | Defect in androgen action | 46,XY —Autosomal rec. | Testis with spermatogenesis | Wolffian | Female | No gynecomastia Nl. testosterone Nl. virilization | None | None | None | |
Complete androgen Insensitivity syndrome | Androgen receptor defect | 46,XY —X linked | 1 in 20,000–64,000 | Testis not fertile | Absent | Female reared as female | Inc. testosterone Inc. estrogen | None | Germ cells | Remove gonads after puberty Estrogen replacement |
Partial androgen Insensitivity syndrome | Androgen receptor defect | 46,XY —X linked rec. | 1/10th of complete | Testis not fertile | Wolffian | Female Versus severe hypospadias | Inc. testosterone Inc. estrogen | None | Germ cells | Depends on sex of rearing |
Infertile male syndrome | Androgen receptor defect | 46,XY —? X linked rec. | Testis not fertile | Wolffian | Male | Infertility Nl. or inc. testosterone Nl. or inc. estrogen | None | None | None | |
Persistent Müllerian duct syndrome | Persistent Müllerian ducts | Unknown | Testis | Wolffian with rudimentary uterus and tubes | Male usually cryptorchid | Nl. testosterone Nl. estrogen | None | None | Orchiopexy Leave uterus and tubes | |
Clinical Evaluation of Patients with Ambiguous Genitalia
The accurate diagnosis of a patient with ambiguous genitalia is a challenging process. Based on the diagnosis, decisions will be made for gender assignment, which will have a great impact not only on the patient but also on the patient’s family (Daaboul and Frader, 2001; Morland, 2001; Conte and Grumbach, 2007). In most societies, the accepted norm is two sexes, either male or female. When a new baby arrives and the proclamation as to whether it is a boy or a girl cannot be made immediately, an anticipated celebration turns into a stressful family dilemma. With prenatal amniocentesis and routine ultrasound, sex determination is often known well before birth. This can compound the emotional trauma when the known and anticipated genotype does not match the newborn’s phenotype. Furthermore, in cases such as severe salt-wasting congenital adrenal hyperplasia (CAH), accurate diagnosis is lifesaving.









