Chapter 12 Congenital Anomalies of the Female Reproductive Tract
INTRODUCTION
In contrast, when congenital anomalies involve either ovaries or external genitalia, the müllerian-derived structures are often normal. In these cases, identifiable aberrations of the external genitalia are often noted in early infancy, with delays in secondary sexual characteristics occurring in later life. specific congenital anomalies of the reproductive tract can be difficult to diagnose because of the wide variety of clinical presentations. Once an accurate diagnosis is rendered, many treatment options exist and are often individualized for a given malformation. For infants born with ambiguous genitalia, a multidisciplinary approach is often necessary. Refinements in surgical and medical techniques have enabled many women with these disorders to have normal satisfying sexual relations. Advances in reproductive technologies have resulted in improved fertility and obstetrical outcomes. Indeed, assisted reproductive technologies now allow many women with congenital anomalies of the reproductive tract to conceive and deliver healthy babies.1
EMBRYOLOGY OF THE FEMALE REPRODUCTIVE TRACT
In the normal female embryo, a series of well-orchestrated, complex interactions is required for proper differentiation of the müllerian ducts and urogenital sinus. Although originating from different germ layers, the fates of the müllerian ducts (mesoderm) and urogenital sinus (endoderm) are interconnected as they differentiate to form the female reproductive tract. The müllerian ducts are the primordial anlage of the internal female reproductive organs and differentiate to form the fallopian tubes, uterus, cervix, and superior aspect of the vagina.1 When the dynamic processes of differentiation, migration, fusion, and canalization are interrupted, a wide spectrum of congenital anomalies of the reproductive tract can result. The anatomic malformations can range from agenesis of the uterus and vagina to duplication of the reproductive organs. Disruption of local mesoderm development and its contiguous somites can account for associated urologic and axial skeletal abnormalities, respectively, that quite often coexist.
Development of the Fallopian Tubes, Uterus, and Uterine Cervix
Two sets of paired genital ducts, paramesonephric (müllerian) and mesonephric (Wolffian), are present in female and male embryos at 6 weeks of development (Fig. 12-1). We use the two terms that refer to each duct somewhat interchangeably. For example, although most embryology books use the term paramesonephric duct, most clinicians use the term müllerian. Development of the mesonephric ducts precedes that of the paramesonephric ducts, and for a short period, the mesonephric ducts drain the contents of the primitive mesonephric kidney into the cloaca.2 A key gene for the development of paramesonephric and mesonephric ducts is PAX2. Mutations in this gene will result in impaired duct and renal development in both sexes.
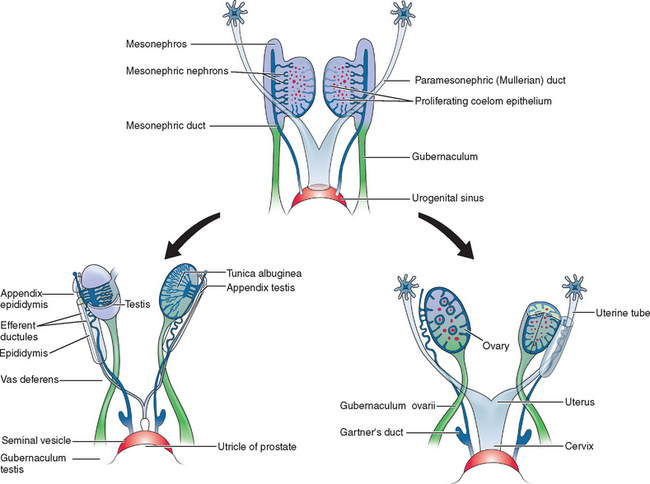
In the female, the mesonephric ducts will degenerate in the absence of testosterone, and the paramesonephric ducts develop because of the absence of anti-müllerian (AMH).2–4 At the same time, the paramesonephric ducts, which originated as longitudinal invaginations of coelomic epithelium, begin to elongate bidirectionally along the anterolateral aspects of the gonads.2 The regressing mesonephric ducts provide an ideal template for the elongating paramesonephric ducts. This primordial connection explains the frequent associations seen later between paramesonephric duct anomalies and urinary system malformations. In the male the paramesonephric ducts will regress under the influence of antimüllerian hormone secreted by the Sertoli cells of the testes. Persistence of the paramesonephric ducts in the male is seen with a mutation of the gene coding for antimüllerian hormone or its receptor.
As the paramesonephric ducts elongate at week 9, three recognizable regions can be identified: the cranial, horizontal, and caudal aspects. Each of these regions will have a distinct fate.2,5 The funnel-shaped cranial regions open directly into the early peritoneal cavity and will form the fimbria of the fallopian tubes. The paramesonephric ducts at this level are lateral to the mesonephric ducts (see Fig. 12-1).
The paired horizontal segments migrate lateral to the mesonephric ducts, then cross ventrally and extend caudomedially to form the remainder of the fallopian tubes. The caudal regions come into contact with their contralateral component at the median plane in the future pelvis and fuse, forming a single Y-shaped tubular structure known as the uterovaginal primordium. The uterovaginal primordium consists of a uterine and vaginal region.2,5 The uterine region gives rise to the uterus; correspondingly, the vaginal component will develop into the superior aspect of the vagina.2,3,5
The uterus has a bicornuate appearance at this stage, but its anatomic configuration will continue to develop through the processes of fusion and subsequent canalization. Uterine septum canalization or regression is mediated by apoptosis, which is regulated by the bcl-2 gene.6 Fusion was thought to occur from a caudal to cranial direction. However, anomalies found in postnatal life, such as a double cervix and vagina but a normal uterus, suggests that fusion may occur initially at the level of the internal uterine isthmus and then proceed in both directions.3
By week 12, the uterine fundus assumes its mature shape. The uterine endometrium is derived from the lining of the fused paramesonephric (müllerian) ducts, and the endometrial stroma and myometrium are derived from adjacent mesenchyme.4,7 This entire process is completed by week 22 of development and results in a uterine cervix and uterus with a single uterine cavity.2
Development of the Vagina and Hymen
Normal vaginal development requires the fusion of two different embryologic structures, the mesodermal paramesonephric (müllerian) ducts and the endodermal urogenital sinus. After formation of the Y-shaped uterovaginal primordium, the caudal tip of the uterovaginal primordium inserts into the dorsal wall of the urogenital sinus (see Fig. 12-1). This event creates an elevation called the müllerian or sinus tubercle. The sinus tubercle induces the formation of the sinovaginal bulbs at its distal aspect. As paired endodermal evaginations, the sinovaginal bulbs extend as a solid core from the urogenital sinus to the caudal aspect of the uterovaginal primordium and then fuse to form the vaginal plate.2,7,8
The vaginal plate and the sinovaginal bulbs restructure the urogenital sinus from its original long, tubular structure into a flat vestibule. This restructuring is responsible for positioning the female urethra at the primitive perineum. Meanwhile, the vaginal plate canalizes caudal to cephalad; when this process is complete at week 20, the vaginal canal is formed. The precise boundary between the uterovaginal primordium and urogenital sinus, as well as their individual contributions to the vagina, has not been established. Some authorities maintain that the superior one third of the vaginal epithelium is derived from the uterovaginal primordium and the inferior two thirds from the uterogenital sinus. However, most other experts consider the entire vaginal lining to be derived from the vaginal plate of the uterogenital sinus.2,7 The vaginal fibromuscular wall develops from the surrounding mesenchyme.
The hymen is formed by expansion of the caudal aspect of the vagina with the subsequent invagination of the posterior wall of the urogenital sinus.2,7 It serves to separate the vaginal lumen from the urogenital sinus cavity until late in fetal development. It ruptures perinatally, with the remnants remaining as a thin mucous membrane.8
Development of the Ovary
Chromosomal and genetic sex is determined at fertilization when the X ovum is fertilized by either an X-bearing or Y-bearing sperm. The XX or XY chromosomal complement of the embryo provides the genes coding for a group of transcription factors—Wilms’ tumor suppressor gene (WT1), DAX1, and steroidogenic factor (SF-1)—that orchestrate gonadal differentiation into either an ovary or testis. WT1 is also important for renal development. Renal and gonadal agenesis will occur with a mutation in the WT1 gene. SF1 and DAX1 gene mutations are associated with gonadal dysgenesis and impaired development of the adrenal cortex. DAX1, an X-linked molecule, suppresses testicular differentiation. Therefore, patients with a mosaic karyotype such as 45,XO/46,XY may have sufficient expression of DAX1 to suppress testicular development. The same concept applies to XXY Klinefelter’s syndrome. Sex determination of the gonads depends on a number of complex molecular events that direct the interactions of the genes encoding these factors.9
Just as the Y chromosome influences the development of the male gonad and phenotype, the X chromosome similarly influences ovarian differentiation and phenotype. The presence of two X chromosomes is crucial to maintenance of normal ovarian function after differentiation. Ovaries of 45,X females demonstrate accelerated oocyte atresia.10 Autosomal genes also influence ovarian development.11
By week 6, the primordial germ cells have integrated into the mesenchyme to become part of the primary sex cords. As the primary sex cords regress, the primordial germ cells then become incorporated into the secondary sex cords, which connect to the ovarian epithelium. At week 12, the ovarian cortex can be identified. Approximately 1 week later, the secondary cords fragment, leaving the primordial follicles.10 The primordial follicle is composed of a primordial germ cell surrounded by one layer of sex cord-derived follicular cells; it is now known as the oogonium. The oogonia undergo active mitosis and initiate meiosis during fetal development. Many degenerate before birth, and only about 2 million will remain after birth; they will enlarge to become primary oocytes.
Development of the External Female Genitalia
The external genitalia of female and male are similar at the indifferent stage of development between weeks 4 and 7 (Fig. 12-2). Distinguishing sexual characteristics begin to appear at week 9, although full differentiation is not achieved until week 12.2 Mesenchyme at the cranial aspect of the cloacal membrane proliferates, forming the genital tubercle. The genital tubercle elongates to form the phallus and later, the clitoris. The genital folds and the labioscrotal swellings develop bilaterally along the cloacal membrane.
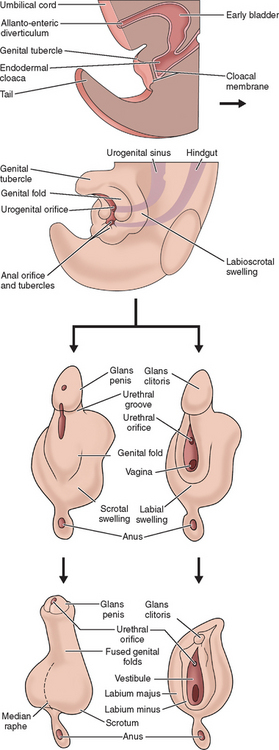
At the completion of week 6, the urorectal septum fuses with the cloacal membrane, dividing the membrane into anal (dorsal) and urogenital (ventral) aspects.2,8 The urogenital membrane lies in the floor of the urogenital groove and is bound by the genital folds. Approximately 1 week later, the membranes rupture to form the anus and urogenital orifices, respectively. The urogenital orifice in the female is the primordial urethra and vaginal vestibule.
The genital folds meet posteriorly, fuse, and form the labia minora frenulum. Their unfused anterior counterparts become the labia minora (minus) proper. The labioscrotal folds also fuse posteriorly and form the posterior labial commissure.2 When they fuse anteriorly, the anterior labial commissure and mons pubis are formed. However, most of the labioscrotal folds remain unfused and form the labia majora.
CLASSIFICATION OF CONGENITAL ANOMALIES OF THE INTERNAL REPRODUCTIVE TRACT
Müllerian anomalies have been reported dating as far back as the 16th century.12 The epidemiology of these anomalies has been vastly outpaced by the remarkable technical achievements used in their diagnosis and treatment. The actual incidences of these anomalies are not well understood. Indeed, reports widely vary, with most authors reporting an incidence ranging from 1:200 to 1:600 in fertile women.13–15
The genetics of various congenital anomalies of the reproductive tract are quite complex and beyond the scope of this chapter. Briefly, most cases occur sporadically. In familial cases, many anomalies appear to be multifactorial. Associations with other modes of inheritance also exist and include autosomal dominant and autosomal recessive patterns of inheritance as well as X-linked disorders. Müllerian anomalies can also represent a component of a multiple malformation syndrome.5,16,17,
CLASSIFICATION SYSTEM FOR REPRODUCTIVE TRACT CONGENITAL ANOMALIES
In 1988, the American Society for Reproductive Medicine (ASRM), formerly known as the American Fertility Society (AFS), published their classification scheme for female reproductive tract anomalies (Fig. 12-3).18 Their classification represents the most widely accepted method of categorizing these abnormalities and has the major advantages of allowing for standardization of reporting methods and providing a template for long-term studies of reproductive outcomes for each anomaly. The system is organized into seven classes according to the major developmental failure and separates the defects into groups having similar clinical manifestations. It also includes a class characterizing uterine abnormalities related to in utero DES exposure. Although vaginal anomalies are not included, the scheme allows for their incorporation.

Figure 12-3 Diagram from the American Fertility Society Classification of Müllerian Abnormalities
(American Fertility Society, Fertil Steril 49:944–955, 1998.)
Class I: Agenesis or Hypoplasia: Segmental or Complete
Agenesis or hypoplasia may involve the vagina, cervix, fundus, fallopian tubes, and/or any combination. The Mayer-Rokitansky-Küster-Hauser syndrome is the most common example in this category.
Class V: Septate Uterus: Complete or Partial
Either a complete or partial midline septum is present within a single uterus.
PATHOGENESIS AND DIAGNOSIS OF DEVELOPMENTAL ANOMALIES OF THE REPRODUCTIVE TRACT
Vaginal Agenesis
Description and Pathogenesis
Vaginal agenesis is the most common defect involving the vagina and uterus and is characterized by the absence or hypoplasia of the uterus and proximal vagina and, in some cases, the fallopian tubes. Frequently, this malformation is accompanied by urinary tract anomalies. Vaginal agenesis occurs when the sinovaginal bulbs fail to develop because the vaginal plate cannot form in the absence of the sinovaginal bulbs.2 It occurs in an estimated 1:5000 newborn females.19 Multiple variants have been reported, with some exhibiting complicated associated anomalies. Although the uterus is usually absent, 7% to 10% of cases will have a normal, albeit obstructed, uterus or a rudimentary uterus with functional endometrium.20 In the presence of active endometrium, the patient may experience cyclical pain.
Vaginal agenesis can be partial or complete. Partial vaginal agenesis is less commonly seen and is characterized by a normal uterus with a small vaginal pouch located distal to the cervix. Complete vaginal agenesis, known as the Mayer-Rokitansky-Küster-Hauser syndrome, is more common. In 90% to 95% of reported Mayer-Rokitansky-Küster-Hauser cases, the uterus is also absent.20–22 The fallopian tubes are normal and the ovaries demonstrate normal endocrine function.
Associated anomalies often coexist with vaginal agenesis. The incidence of associated urologic abnormalities varies from 15% to 40%; skeletal anomalies such as congenital fusion or absence of vertebra occur in approximately 12% to 50%.20,23 An association between the Mayer-Rokitansky-Küster-Hauser syndrome and the Klippel-Feil syndrome has been reported. Components of the Klippel-Feil syndrome include congenital fusion of the cervical spine, short neck, a low posterior hairline, and limited range of motion in the cervical spine.24 The Mayer-Rokitansky-Küster-Hauser is also associated with the MURCS syndrome. MURCS syndrome is characterized by aplasia of the müllerian ducts and renal system as well as cervicothoracic somite dysplasia.25,26
Patients with vaginal agenesis are 46,XX. Most cases occur sporadically, although approximately 4% of reported cases are clustered in families.20,21 Abnormalities in the expression of homeobox (Hox) genes are associated with müllerian duct agenesis. It has been reported that increased exposure to galactose is responsible for abnormal vaginal development based on the finding that vaginal agenesis has been reported to be associated with variants of the galactose 1-phosphate uridyl transferase enzyme.27–29 Other authorities have speculated that mutations in either the antimüllerian hormone or müllerian inhibitory substance gene or its receptor gene are responsible for this disorder.4 In a recent report, a de novo translocation in a young woman with Mayer-Rokitansky-Küster-Hauser syndrome is discussed, suggesting that this break point may be involved in midmüllerian differentiation.30
Diagnosis of Vaginal Agenesis
Vaginal agenesis is usually diagnosed at puberty when young adolescents present with primary amenorrhea. Indeed, vaginal agenesis is the second most common cause of primary amenorrhea in adolescents.20,21 Normal growth and development and the presence of age-appropriate secondary sexual characteristics are all evident. The external genitalia have a normal appearance, although visual inspection can often reveal a patulous urethra.30,31 The appearance of the vagina can be diverse; it can be completely absent or can be present as a short, blinding-ending pouch or as a vaginal dimple. The appearance of the vaginal dimple can range from a slight indentation to up to 5 to 6 cm in length. A uterus is not palpated on rectal examination.
Pelvic ultrasonography can support the clinical findings of an absent uterus or uterine remnant and the presence of normal ovaries. Magnetic resonance imaging (MRI) is extremely useful for those cases in which ultrasound findings are not definitive; lack of visualization of the vagina and a uterus in a technically adequate study indicates agenesis or hypoplasia of these structures.32–34 Laparoscopy is not usually indicated unless the diagnosis cannot be determined by other studies or if there is concern over the presence of a functioning uterus or rudimentary uterine tissue.
Unicornuate Uterus
Description and Pathogenesis
A unicornuate uterus occurs when one of the paired müllerian ducts fails to elongate while the other develops normally. This anomaly accounts for approximately 13% of all müllerian duct abnormalities.35 These ASRM class II uterine defects are structurally quite diverse. The unicornuate uterus may occur in isolation but is frequently accompanied by a rudimentary horn.35,36 Associated urologic anomalies frequently (44%) occur, especially when the rudimentary horn is obstructed. These anomalies include ipsilateral renal agenesis (67%), horseshoe kidneys, or ipsilateral pelvic kidney (15%).35,36
Obstetric outcomes are generally poor, although normal pregnancies can occur. Because these anomalies are uncommon, most reports do not classify reproductive outcomes according to the various malformation subclasses.35 For the entire unicornuate class, the spontaneous abortion rate is 51%, the preterm birth rate is 15%, and the fetal survival rate is approximately 40%.37 Common obstetrical complications include malpresentation, intrauterine growth retardation, and preterm birth.37–41
Diagnosis of Unicornuate Uterus
Although a hysterosalpingogram (HSG) is useful in diagnosing a unicornuate uterus, it cannot detect a noncommunicating rudimentary horn. MRI can reliably identify a noncommunicating horn and is characteristically not opacified when the endometrium is absent.32 High-resolution ultrasound evaluation can usually identify rudimentary horns and is more reliable than laparoscopy in determining whether the horn is communicating. Indeed, laparoscopy is rarely indicated as part of the diagnostic evaluation of an obstructed, noncommunicating horn. Additional studies should include an intravenous pyelogram (IVP) or renal ultrasound to evaluate for ipsilateral renal agenesis, a horseshoe kidney, or an ipsilateral pelvic kidney.35,42
Uterus Didelphys
Pathogenesis
Either unilateral or bilateral duplication of the müllerian ducts can result in uterus didelphys. This anomaly is characterized by the presence of two separate, normal-sized uteri and cervices that are fused at the lower uterine segment. Vaginal duplication is frequently a component, and an associated vaginal septum is often seen. A longitudinal (horizontal) septum extends either completely or partially from the cervices to the introitus. A complete vaginal septum with a sagittal orientation occurs in approximately 75% of cases; some have occasional obstructing transverse septum.43–45
Uncommonly, patients can present with an obstructed unilateral vagina. This abnormality is frequently associated with ipsilateral renal and ureter agenesis and is a component of the Wunderlich-Herlyn-Werner syndrome.46,47
Reported reproductive outcomes are slightly better for uterus didelphys than for the unicornuate uterus. Fertility is not compromised, but the spontaneous abortion rate is high (40%). However, when the pregnancies are carried to term, few obstetrical difficulties occur.35,37,38
Diagnosis of Uterus Didelphys
Uterus didelphys with obstructed unilateral vagina can be diagnosed early and accurately. The clinical presentation is similar to that in patients having a unicornuate uterus and noncommunicating, functional horn.35,38 Ipsilateral renal agenesis frequently accompanies both these conditions as well.46,47 On pelvic examination, one cervix is identified, and a paravaginal cystic-type structure can usually be palpated that represents the noncommunicating second vagina.
Preoperative diagnostic studies are similar to those used for the unicornuate uterus and include HSG, pelvic ultrasound or MRI, and an IVP or renal ultrasound to either confirm or exclude associated urinary tract anomalies.33,34,42
Bicornuate Uterus
Pathogenesis
Müllerian ducts that incompletely fuse at the level of the uterine fundus result in the bicornuate uterus malformation. The lower uterus and cervix are completely fused, but the incompletely fused ducts elongate and develop into two separate uterine horns. Their respective endometrial cavities communicate, although an intervening muscular uterine septum is present. A single-chamber cervix and vagina are present. The subclassification of this anomaly is contingent on whether the separation is complete or partial. Complete variants include the uterus bicornis unicollis, in which the uterine cavity division extends to the internal os, and the uterus bicornis bicollis, in which the division extends to the external os. Partial bicornuate uteri are characterized by a uterine division that is confined to the fundal region. In this anomaly, the uterine cavity division corresponds to a visible sagittal groove on the external fundal uterine surface. The depth of the groove and the extent of the separation are subject to the length of the incompletely fused müllerian ducts.35,48
Usually the bicornuate uterus is an incidental finding. Women with this uterine malformation usually have no difficulty becoming pregnant, and approximately 60% can expect to deliver a viable infant.36 However, they can present with late abortion or premature labor. This observation may be related to whether the bicornuate uterus is partial or complete. According to one study, women with partial bicornuate uterus experienced a spontaneous abortion rate of 28% and preterm delivery rate of 20%. These rates contrasted with a spontaneous abortion rate of 66% and a higher rate of preterm deliveries in women diagnosed with a complete bicornuate uterus.35
Diagnosis of Bicornuate Uterus
Evaluation begins with an ultrasound performed during the luteal phase of the menstrual cycle when the endometrial echo complex is better identified.42 Ultrasound based on the angle between the two cornu cannot accurately distinguish the bicornuate uterus from the septate uterus, and MRI must be performed to establish this diagnosis. HSG, usually the cornerstone in diagnosing most uterine structural anomalies, cannot reliably distinguish between these two entities because they can display similar uterine cavity images.49,50
Septate Uterus
Pathogenesis
There are three types of septa: complete, partial, and segmental. The complete septum extends from the fundus to the internal os, dividing the endometrial cavity. The partial septum is located at the fundus; the segmental septum, also located at the fundus, is noncontiguous, allowing for partial communication between the endometrial cavities.51,52
The fertility of women with this type of anomaly is not significantly compromised. However, the septate uterus is associated with the poorest reproductive outcomes of all the müllerian duct anomalies. Spontaneous abortion rates of 67% have been reported, and the live birth rate ranges from 15% to 28%; obstetric complications such as incompetent cervix, premature labor, and abnormal presentation are common.53–56 Hysteroscopic resection of the septum increases the live birth rate to as high as 75%.
Diagnosis of the Septate Uterus
HSG is an essential study in diagnosing and planning surgery for the septate uterus. It can assess the presence of a two-chambered uterus, determine the length and thickness of the septum, and permit concomitant assessment of tubal patency. However, HSG is limited since it cannot distinguish the septate from the bicornuate uterus or detect minor septal defects.50 MRI provides excellent tissue characterization and can reliably differentiate between septate uterus and bicornuate uterus.56 Transvaginal ultrasound can also be useful in identifying this abnormality. In one study, transvaginal ultrasound had a sensitivity of 100% and a specificity of 80% in detecting the septate uterus.57 Additionally, if three-dimensional ultrasound is available, the reported accuracy was 92% for the diagnosis of septate uterus.
Arcuate Uterus
Description
The arcuate uterus has a small (<1.5 cm) septate projection located at the fundal aspect of the uterine cavity. The external contour of the uterus is convex or flat. This congenital anomaly is the most common uterine abnormality detected using HSG.58,59 Classifying the arcuate uterus has been challenging. Earlier classification systems considered it a mild form of the bicornuate uterus and the ASRM classification places this anomaly in a separate class.18 Some speculate that the arcuate uterus represents a normal variant because it is not associated with an increased risk of spontaneous abortion or other pregnancy complications. Interestingly data on outcome after resection of the septum in a septate uterus demonstrates that leaving approximately 1 cm of septum will not affect pregnancy outcomes.
Diagnosis of Arcuate Uterus
There is limited data regarding the diagnosis, management, and reproductive outcomes for the arcuate uterus. Using HSG, a single uterine cavity with a saddle-shaped fundus can be identified. MRI shows a normal fundal contour with a minimal indentation.58,59 Renal ultrasound and IVP may be performed to exclude any associated urinary tract anomalies, although these studies generally are not part of a standard evaluation. Management is similar to the septate uterus with only selected patients fulfilling criteria for poor reproductive performance recommended for surgical correction.
Diethylstilbestrol-Related Anomalies
Pathogenesis
DES is a synthetic estrogen prescribed from the late 1940s to the 1970s to prevent recurrent pregnancy loss, premature delivery, and other obstetric problems. In the early 1970s it was found to be a teratogen and its use during pregnancy was banned.60 It is now known that there is a strong association between in utero DES exposure and an increased risk of developing vaginal adenocarcinoma later in life. Additional studies revealed that in utero DES exposure was also associated with structural abnormalities of the developing uterus, cervix, and vagina. Although some women exposed to DES in utero are in their midthirties, the majority of these women are now postmenopausal or approaching the end of their reproductive years.
Uterine anomalies include a T-shaped endometrial cavity, widened lower uterine segment, midfundal constrictions, endometrial filling defects, and uterine hypoplasia.61,62 DES-associated structural abnormalities of the cervix also have been identified but are less frequent than those of the uterus. These abnormalities include cervical hypoplasia, anterior ridge or collar (cock’s comb cervix), and pseudopolyps. Vaginal adenosis and vaginal constrictions have also been associated with DES.61,62 Interestingly, uterine anomalies similar to those related to DES exposure have been reported in women without in utero DES exposure.
Although there is no convincing evidence that in utero DES exposure has an unfavorable impact on fertility, there is considerable evidence to indicate that many patients with DES exposure will have poor obstetrical outcomes. Complications include an increased risk of spontaneous abortions, ectopic pregnancies, and cervical incompetence.63 However, it remains uncertain whether the anomalies themselves or some subclinical abnormality of these women’s reproductive tracts are responsible for these associations.






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


