CHAPTER 123 Colorectal Cancer
Cancer of the colon and rectum (colorectal cancer [CRC]) is a major cause of cancer-associated morbidity and mortality in North America, Europe, and other regions with similar lifestyles and dietary habits. CRC is the fourth most common newly diagnosed internal cancer overall in the United States, after cancers of the lung, prostate, and breast, and currently constitutes 10% of new cancers in men and women. In 2009, there were an estimated 147,000 new CRC cases in the United States and 50,000 related deaths (a rate second only to that of lung cancer).1 In the United States, CRC incidence rates in men and women are similar, although there appears to be a slight male predominance worldwide. Approximately 6% of the American population eventually will develop invasive colon or rectal cancer, and more than 6 million Americans who are alive today will die of the disease; the lifetime risk of dying from CRC in the United States is 2.5%. Globally, CRC is the fourth most common cancer in men and the third most common in women, with mortality paralleling incidence. Despite evidence that five-year survival is 90% when CRC is diagnosed at an early stage, less than 40% of cases are diagnosed when the cancer is still localized.2
EPIDEMIOLOGY
The frequency of CRC varies remarkably among different populations (Fig. 123-1).3 Incidence rates are highest in the developed countries in North America and in Australia and New Zealand; intermediate in Europe; and low in Asia, South America, and especially sub-Saharan Africa. Internationally, the incidence of colon cancer in men differs by a factor of almost 90 between areas with the extreme lowest and highest rates; the incidence of rectal cancer (cancer within 11 cm of the anus) differs by a factor of 13. CRC incidence also differs within countries, depending on region and population (Fig. 123-2). These differences most likely are due to differences in environmental factors, including dietary patterns (discussed later).
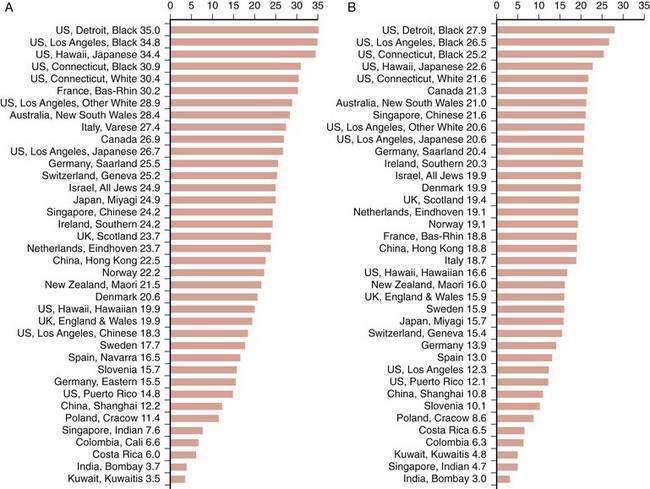
Figure 123-1. Age-standardized incidence of colon cancer per 100,000 population in various populations for men (A) and women (B).
(Data from Parkin DM, Whelen SL, Ferlay J, et al. Cancer incidence in five continents. [IARC Sci. Publ. No. 143]. Lyon: International Agency for Research on Cancer; 1997; and Curado MP, Edwards SB, Shin HR, et al. Cancer incidence in five continents, vol. IX [IARC Sci. Publ. No. 160]. Lyon: International Agency for Research on Cancer; 2007.)
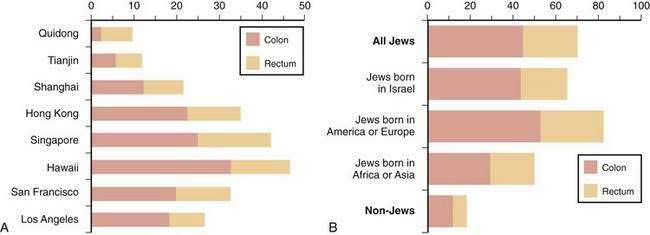
Between 1950 and the mid-1980s, the incidence of colon cancer in the United States rose in the white population, whereas that of rectal cancer remained fairly stable. Mortality rates from CRC were stable among white men but decreased in white women. Both incidence and mortality rates for CRC increased substantially among the nonwhite population during this period. CRC incidence and mortality have declined since 1985 in American adults at an average annual rate of 1.6% and 1.8%, respectively2; these trends are more evident in whites than blacks. Overall death rates from CRC declined between 1990 and 2004 by almost 30% in men and 25% in women.1 Currently, incidence and mortality rates for CRC are higher in the African American population compared with the white population, and these rates in the U.S. Latin American population are slightly lower.1
The risk of CRC rises rapidly in populations that migrate from areas of low risk to areas of high risk. This pattern was demonstrated clearly in Japanese immigrants to Hawaii and to the continental United States during the 1950s and 1960s. Cancer rates for Issei (the migrating generation) rose over a short period to exceed those of native Japanese living in Japan, and the incidence rates for Nissei (their U.S.-born offspring) rose progressively to approximate those of the native white population. A similar upward displacement of CRC risk was noted in Europeans who migrated to Australia after World War II and in Jews who migrated to Israel from low-risk areas in Yemen and North Africa. Longitudinal studies reveal that in many countries where CRC mortality rates were low before 1950, rates have increased sharply, whereas in countries where rates were high or moderate, they have decreased, stabilized, or increased slightly. Japan is a good example of this change: Once a low-risk region for colon cancer, incidence rates have risen to equal or exceed those in North America and Europe.3
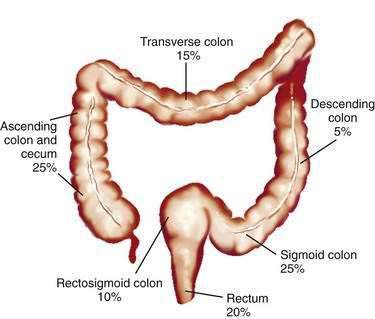
Studies of temporal trends by subsite location of large bowel cancer demonstrate that for both sexes, incidence rates have increased for cancers of the right colon (cecum, ascending colon) and sigmoid colon and have decreased for cancers of the rectum; this change might reflect differing susceptibilities to neoplastic transformation in the proximal and distal colon. Currently in the United States, the prevalence of CRCs in whites is higher in the cecum and ascending colon (22% in men, 27% in women) and in the sigmoid colon (25% in men, 23% in women) than elsewhere in the large bowel (Fig. 123-3).
ETIOLOGY
Inter-regional differences in the incidence of CRC, including differences among population groups living in geographic proximity but with different lifestyles, strongly suggest that environment plays a role in the development of this disease. Migrant studies and rapid changes in incidence in countries assimilating Western practices support this concept. Strong circumstantial evidence exists for a link between diet and CRC. Population studies and animal studies have attempted to delineate the effects of various fats and proteins, carbohydrates, vegetable and fiber components, and micronutrients on the genesis of cancer of the large bowel (Table 123-1).
Table 123-1 Factors That May Influence Carcinogenesis in the Colon and Rectum
| Probably Causative |
| Possibly Causative |
| Probably Protective |
| Possibly Protective† |
NSAIDs, nonsteroidal anti-inflammatory drugs.
* Dietary fats and fiber are heterogeneous in composition, and not all fats or fiber components play a role in cause or protection.
FAT, BILE ACIDS, AND BACTERIA
Activation of protein kinase C (PKC) by bile acids in colonic mucosa also might represent an important intracellular event by which bile acids provoke a proliferative response. Mucin alterations are a common feature of colonic neoplasia, and alterations in MUC2 mucin have been associated with tumor progression in the colon. Bile acids induce mucin expression in human colon carcinoma cells by increasing MUC2 transcription through a process involving mitogen-activated protein (MAP) kinase-independent, PKC-dependent activation of the transcription factor AP-1.4 Bile acids can, in addition, induce release of arachidonate and conversion of arachidonic acid to prostaglandins in the mucosa, which can enhance cell proliferation.5 Preclinical and clinical evidence indicate that nonsteroidal anti-inflammatory drugs (NSAIDs), which reduce prostaglandin synthesis, reduce the incidence of large bowel cancer (discussed later); inhibition of the inducible enzyme cyclooxygenase (COX)-2 may be particularly important in this regard.6
An inverse relationship has been reported between physical activity and risk for CRC in men; obesity is associated with elevated risk of CRC. Serum cholesterol and β-lipoprotein levels have been positively correlated with the development of colorectal adenomas and CRC, but this association has not been demonstrated consistently, and serum cholesterol levels can decline before the development of colon cancer. Adiponectin is a hormone secreted by adipose tissue, serum levels of which are inversely correlated with obesity and hyperinsulinemia. Variants of adiponectin and adiponectin receptor genes have been correlated with differences in CRC risk.7
FIBER
Epidemiologic, case-control, and animal studies suggest that dietary fiber protects against the development of colon cancer. Dietary fiber is plant material that resists digestion and that is composed of a heterogeneous mix of carbohydrates (e.g., cellulose, hemicellulose, pectin) and noncarbohydrates (e.g., lignin). Although the protective role of fiber is not completely clear, epidemiologic studies correlate high-fiber intake with a lower incidence of colorectal neoplasia.8,9 The majority of observational-epidemiologic and case-control studies support the protective effect of fiber-rich diets; however, these data do not define the relationship between fiber-rich food and the importance of nonfiber vegetable components, nutrients, and micronutrients in fruits and vegetables. The effect of fiber components on different portions of the large bowel also can vary. This might explain, in part, the inability to demonstrate a protective effect of fiber in several randomized, controlled trials that have examined the ability of fiber supplementation to prevent adenoma recurrence.10,11
Two large prospective observational studies, including one from the Prostate, Lung, Colon and Ovarian Cancer Trial (PLCO) found that increased fiber intake is significantly associated with reduced risk of colorectal neoplasia.8 Another analysis of 13 prospective cohort studies included in the Pooling Project of Prospective Studies of Diet and Cancer demonstrated that dietary fiber intake was inversely associated with risk of CRC in age-adjusted analyses, but that after accounting for other dietary factors, high intake of dietary fiber was not associated with a reduced risk of CRC.12
CALCIUM AND VITAMIN D
Epidemiologic, clinical, and laboratory evidence suggest that calcium intake might protect against carcinogenesis in the colon. Calcium has numerous biological effects that might reduce colon carcinogenesis, including actions on the cell cycle, cyclic adenosine monophosphate (cAMP), calmodulin, tyrosine kinases, ornithine decarboxylase, and e-cadherin. The calcium-sensing receptor expressed in the intestine senses extracellular calcium with effects on differentiation and proliferation. The potential chemopreventive activity of calcium was suggested originally by epidemiologic studies reporting an inverse relationship between CRC and intake of vitamin D and calcium. Dietary calcium supplementation in the form of low-fat dairy foods can affect a variety of intermediate biomarkers thought to be associated with tumor progression in the colon, and supplemental calcium plus vitamin D alters preneoplastic features of colorectal adenomas.13 Although the relationship between calcium intake and a lower incidence of colonic adenomas and carcinomas has not been uniformly demonstrated, observational studies overall suggest a protective effect. A pooling project of 10 cohort studies strongly suggested that calcium intake is inversely related to the risk of CRC.14
Vitamin D3 metabolites and analogs have been shown to play an important role in the regulation of a number of important cellular processes, including proliferation, differentiation, and apoptosis, in addition to their established role in mineral homeostasis. These steroid compounds have rapid effects that do not involve gene transcription or protein synthesis, as well as genomic effects involving the vitamin D receptor and other transcription factors.15,16 Vitamin D can modulate more than 200 genes involved in cell cycle regulation, growth factor signaling, protection against oxidative stress, bile acid and xenobiotic metabolism, cell adhesion, DNA repair, angiogenesis, inflammation, and immune function. Effects of vitamin D and its metabolites have been demonstrated in normal and malignant colonocytes, and several potential mechanisms have been suggested by which these compounds might prevent carcinogenesis in the colon. Dietary supplementation with calcium and vitamin D in rodents fed colon tumor-inducing Western diets significantly reduced tumor incidence and multiplicity in addition to altering expression of a variety of genes linked to initiating formation of colon tumors.17
ARACHIDONIC ACID, EICOSANOIDS, AND CYCLOOXYGENASE-2
This enzyme exists in two isoforms: COX-1 and COX-2. COX-1, the constitutive form of the enzyme, is present in most tissues and is involved in the physiologic production of prostaglandins to maintain normal homeostasis. COX-2 is induced by cytokines, mitogens, and growth factors, and its level has been shown to be elevated in both murine and human CRCs.18–22 Expression of COX-2 is increased markedly in 85% to 95% of CRCs and in experimental models of CRC. COX-2 inhibition prevents cancer from developing during the initiation and the promotion and progression stages of carcinogenesis.21 Knockout of COX-2 results in suppression of intestinal polyposis in animal models of familial adenomatous polyposis (FAP).19 15-Hydroxyprostaglandin dehydrogenase (15-PGDH) is a prostaglandin-degrading enzyme that is lost in human colon cancers and has been shown to be a physiologic antagonist of the prostaglandin-synthesizing activity of COX-2.22 It has been speculated that NSAIDs might reduce formation of colon tumors by inhibiting prostaglandin-mediated proliferation, but other evidence suggests that part of their effect might result from inducing apoptosis. Overexpression of COX-2 has been demonstrated to decrease apoptosis, whereas inhibition of COX-2 leads to an increase in apoptosis.
Other potential mechanisms by which COX-2 inhibition might affect tumor formation include alterations of cell adhesion to extracellular matrix proteins, inhibition of tumor neovascularization (angiogenesis), and reduction in carcinogen activation. A study using the ApcΔ716 mouse, an animal model of FAP, demonstrated that treatment with the COX-2 specific inhibitor rofecoxib (Vioxx) was associated with a significant dose-dependent reduction in size and number of polyps as well as alterations in polyp morphology. COX-2 inhibition was associated with decreased levels of vascular endothelial growth factor (VEGF) and with lower rates of DNA replication.20
CHEMOPREVENTION
Chemoprevention refers to the use of natural or synthetic agents to reverse, suppress, or prevent progression or recurrence of cancer23,24; this is a cornerstone of primary prevention. Data on chemoprevention of CRC come from studies in laboratory animals (see earlier), observational epidemiologic studies, case-control studies, and randomized controlled trials (RCTs). Because the natural history of CRC is protracted, clinical RCTs often have concentrated on preventing colorectal adenomas, which represent a form of intraepithelial neoplasia and are the precursors to carcinoma. The duration of the studies required, sample sizes necessary, cost, and ethical considerations make the use of cancer as an end point impractical. This difficulty has led to an increasing use of surrogate biomarkers to study chemoprevention of CRCs,25 with the hope that use of such markers will lead to shorter, smaller, and less expensive trials.26 To be valid, however, such biomarkers need to accurately represent the events involved in the process of carcinogenesis. When an intervention such as a chemopreventive agent is tested, there should be a clear relationship among the agent, modulation of the biomarker, and the development of cancer. Surrogate end points for cancer ideally should be validated in the context of clinical studies that use cancer as the ultimate end point. This is a difficult task because these are the very trials that such markers are designed to complement or replace.
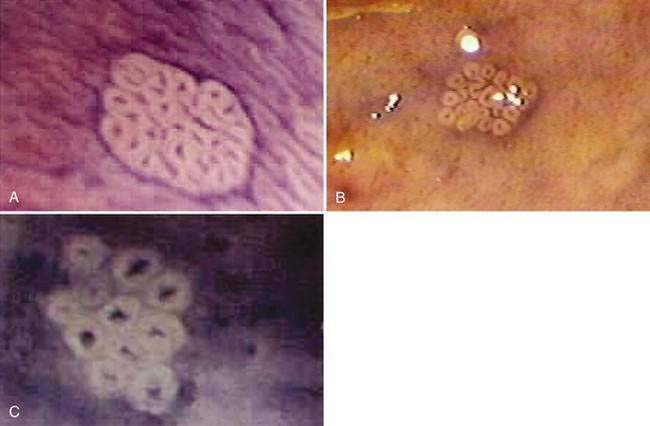
There has been interest in the use of magnifying endoscopy to study aberrant crypt foci of the colon as possible markers in chemoprevention trials.24 These foci consist of large, thick crypts that can be detected by chromoendoscopy using agents such as methylene blue or indigo carmine (Fig. 123-4). Aberrant crypt foci, particularly large crypts with dysplastic features, are thought to be precursors of adenomas in the colon. Standardization of techniques to identify and quantify these lesions is crucial to the successful interpretation of data from these trials, because it is unclear whether these lesions, which appear to be precursors to neoplasia in animal models, play a similar role in the human colon.27 Studies suggest that the majority of aberrant crypt foci in the human colon may be hyperplastic rather than dysplastic (see Chapter 122).
The potential benefit of low-fat, high-fiber diets based on descriptive epidemiology and case-control studies already has been discussed, but current data from prospective human trials are thus far equivocal or negative. Two large RCTs examined the effects of fiber supplementation on recurrence of adenomas. The Polyp Prevention Trial11 randomized 2079 subjects with a history of colorectal adenomas to receive counseling together with a low-fat, high-fiber diet rich in fruits and vegetables or to receive their usual diet alone. The incidence of recurrent adenomas at one and four years, as determined by colonoscopy, was similar in both groups. In a study conducted by the Phoenix Colon Cancer Prevention Physicians’ Network,10 1429 patients with a history of colorectal adenoma were randomized to receive 2.0 g or 13.5 g of supplemental wheat bran per day. Colonoscopy failed to show a difference in the incidence of recurrent adenomas at a median follow-up of 34 to 36 months.
A large body of observational and laboratory studies suggests a role for dietary calcium supplementation in chemoprevention. A prospective double-blind placebo-controlled trial showed that supplemental calcium (3000 mg of calcium carbonate per day, equivalent to 1200 mg of elemental calcium) reduced the incidence and number of recurrent adenomas in subjects chosen for a recent history of such lesions.26 The effect of calcium was modest: 19% reduction in recurrence of adenomas and 24% reduction in the number of adenomas over three years, independent of age, sex, or dietary intake of calcium, fat, or fiber. The protective effect of calcium supplementation on the risk of colorectal adenoma recurrence extended up to five years after cessation of active treatment, even in the absence of continued supplementation.28 Analysis of subjects’ serum vitamin D status suggested that calcium supplementation and vitamin D status appear to act together to reduce the risk of adenoma recurrence.29 The results of a Japanese study, the Fukuoka Colorectal Cancer Study,30 support the joint action of calcium and vitamin D in preventing CRC. Human trials using antioxidant vitamins A, C, and E have provided equivocal results, and current data do not support their routine use for colon cancer prevention in average-risk persons.
Folic acid and its metabolites play an important role in DNA synthesis, strand integrity, and methylation. Epidemiologic studies have found a lower incidence of CRC among those with high compared with low dietary intake of folate.31 This protective effect also was suggested by the Nurses’ Health Study, in which high doses of folate (as part of multivitamin supplementation) given over several years were protective against CRC. A large prospective RCT32,33 failed, however, to demonstrate a protective effect of 1 mg/day of folate supplementation on recurrence of adenoma compared with placebo and suggested that folate supplementation in persons with prior adenomas actually might increase adenoma risk. Analysis of baseline dietary and serum folate levels in these subjects supports the idea that although moderate doses of folate may be protective compared with deficiency, at some point of sufficiency, supplementation provides no additional benefit.34 The lack of efficacy of folate (0.5 mg/day) supplementation to prevent adenoma recurrence also was found in another RCT.35
Epidemiologic, case-control, and prospective cohort trials suggest a protective effect against the development of CRC in women taking hormone (estrogen) replacement therapy. It has been postulated that estrogen might protect against colon cancer by decreasing production of secondary bile acids, by decreasing levels of insulin-like growth factor-1, or through as yet undetermined direct effects on colonic mucosal epithelial cells. Data from the Women’s Health Initiative demonstrated that short-term use of estrogen plus progestin was associated with a decreased risk of CRC; however, CRCs in women who took estrogen plus progestin were diagnosed at a more advanced stage than those in women who took placebo.36
Cigarette smoking has been associated with incidence of and mortality from CRC in observational studies,37 but the long-term effects of smoking cessation on CRC have not been studied.
The most promising results for CRC prevention come from trials using aspirin and NSAIDs. Case-control and cohort studies have suggested that the risk for development of adenoma and carcinoma may be reduced substantially (40% to 50%) among aspirin and NSAID users, compared with controls.6,24,38 A prospective cohort study among male health professionals demonstrated that persons who take aspirin more than twice per week were at lower risk for CRC (relative risk [RR], 0.68) than were controls. An RCT that assessed the effect of low-dose aspirin in an average-risk population demonstrated no significant reduction in the number of CRC cases during the first six years of follow-up; longer follow-up may be necessary to demonstrate a significant effect of aspirin on development of cancer, because the Nurses’ Health Study demonstrated that the benefits of aspirin might not be evident until after at least a decade of regular aspirin consumption.6 Three prospective adenoma prevention trials (see later) now provide compelling evidence that aspirin use reduces the risk of developing colorectal adenoma in persons with a personal history of adenoma or carcinoma.39–41
Related posts:
 Surgical Peritonitis and Other Diseases of the Peritoneum, Mesentery, Omentum, and Diaphragm
Surgical Peritonitis and Other Diseases of the Peritoneum, Mesentery, Omentum, and Diaphragm
 Hepatitis Caused by Other Viruses
Hepatitis Caused by Other Viruses
 Hemochromatosis
Hemochromatosis
 Gastrointestinal and Hepatic Complications of Solid Organ and Hematopoietic Cell Transplantation
Gastrointestinal and Hepatic Complications of Solid Organ and Hematopoietic Cell Transplantation
 Nutritional Assessment and Management of the Malnourished Patient
Nutritional Assessment and Management of the Malnourished Patient
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


