Physical exam findings
Symptoms
RUQ tenderness 2+
Confusion 1+
RUQ abdominal discomfort 2+
Hepatomegaly 2+
Fever 1+
Weight loss/gain 2+
Ascites 2+
Finger clubbing 1+
Anorexia 2+
Malnutrition and wasting 2+
Dupuytren’s contractures 1+
Fatigue 2+
Venous collaterals 1+
Leg edema 1+
Muscle cramps 2+
Splenomegaly 1+
Parotid gland enlargement 1+
Pruritus 1+
Jaundice 1+
Gynecomastia 1+
Nausea and vomiting 1+
Palmar erythema 1+
Testicular atrophy 1+
Confusion/mental disturbance 1+
Cutaneous telangiectasia 1+
Dementia 1+
GI bleeding 1+
Asterixis 1+
Peripheral neuropathy 1+
Sleep disturbance 1+
Patients with compensated cirrhosis are frequently asymptomatic, but they may have anorexia, nausea, weight loss, fatigue, weakness, abnormal menstruation, loss of libido, muscular cramps, and/or difficulty concentrating on mental tasks. Patients with decompensated cirrhosis are often jaundiced, have evidence of muscular wasting, feel weak, and develop fluid retention with edema and abdominal distention. In addition, many complain of itching and others present with hematemesis or melena. Easy bruising, inverted sleep pattern, and confusion are also frequent complaints.
Physical Exam
On physical exam, signs of fatty liver can range from mild hepatomegaly, with blunting of the normally sharp liver edge, to massive hepatomegaly (Table 8.1). In patients with alcoholic hepatitis , hepatomegaly (80–90 % of cases), jaundice (40–60 %), and fever (23–56 %) are very common. If the injury is severe, the patients will have ascites, hepatic encephalopathy, splenomegaly, evidence of muscle wasting, and sometimes gastrointestinal hemorrhage with portal hypertensive gastropathy or gastroesophageal varices [11, 12].
Patients with compensated cirrhosis often have hepatomegaly with hard liver consistency and a nodular surface. They may also have splenomegaly and, less often, right upper quadrant pain. Less common findings include gynecomastia and testicular atrophy [13], amenorrhea, parotid enlargement [14], cutaneous spider angioma [15], Dupuytren’s contractures [16], digital clubbing [17] (found especially in patients with hypoxemia related to hepatopulmonary syndrome), palmar erythema, and nail changes.
Patients with decompensated cirrhosis may also have mental changes of hepatic encephalopathy with variable degrees of confusion, with or without asterixis, jaundice, ascites, and peripheral edema. Some patients will have evidence of gastrointestinal bleeding or other organ damages, including alcoholic gastritis or pancreatitis, alcoholic neuropathy, or, less commonly, alcoholic cardiomyopathy. Signs of alcohol withdrawal are common in patients with alcoholic hepatitis who have recently discontinued alcohol.
Laboratory
Patients with ALD should have a complete blood count, international normalization ratio (INR), comprehensive metabolic panel, and GGT. Because there is the risk of overlapping viral hepatitis, serologies for current infection or past exposure to hepatitis A, B, and C are advised. Patients who are not immune should be vaccinated against hepatitis A and B. If the ALT is elevated, a full workup for other causes of liver disease and cirrhosis is also advisable because other immune or metabolic causes may be uncovered.
Common hematologic findings include anemia, macrocytosis, leukopenia, lymphocytopenia, and thrombocytopenia. Macrocytosis is often due to alcohol toxicity to the bone marrow, but is important to assess for folic acid and/or vitamin B12 deficiency. In patients with alcoholic hepatitis without cirrhosis, thrombocytopenia frequently reverses with rebound thrombocytosis after 1–3 weeks [18]. In cirrhotic patients, thrombocytopenia persists as evidence of portal hypertension. An elevated INR is a marker of the severity of the disease, but can also signal a nutritional deficiency; hence, vitamin K repletion can be helpful in clarifying the situation by eliminating the nutritional deficiency component.
The comprehensive metabolic panel helps to identify electrolyte imbalances that need prompt correction. Similarly, measurements of magnesium, zinc, and phosphorous in plasma can be useful. We frequently supplement with magnesium (magnesium oxide 400 mg/day) and zinc (zinc sulfate 220 mg/day) to treat muscle cramps. Patients with alcoholic hepatitis and with decompensated cirrhosis are very susceptible to kidney injury, and creatinine elevated above the patient’s usual “baseline” requires prompt attention and intervention to avoid progression of acute kidney injury and development of hepatorenal syndrome.
Consumption of alcohol in excess of 50 g per day causes elevation of AST (sensitivity 50 %, specificity 82 %) and ALT (sensitivity 35 %, specificity 86 %) [19]. The elevation of AST is typically higher than that of ALT, and 79 % of patients with alcoholic hepatitis have an AST–ALT ratio >2. Patients with alcoholic hepatitis with ALT > AST frequently have overlapping causes, such as superimposed viral hepatitis or drug injury (most often acetaminophen); the same is true if the AST or ALT is higher than 300 units/L, because this threshold is not likely to be surpassed by alcohol injury alone. Elevated GGT is more sensitive for alcohol abuse (56–73 %) but less specific (53–70 %) than CDT or MCV [20]. Using a combination of these tests may be warranted. Elevated bilirubin, in the absence of biliary obstruction, is a marker of the severity of the alcoholic liver injury and is very important as a component of the Maddrey discriminant function , MELD score, Glasgow alcoholic hepatitis score, the Lille model, and the Child–Turcotte–Pugh calculator index (discussed subsequently).
Validated self-reported questionnaires are superior to biochemical tests in detecting alcohol abuse. Biochemical tests can be utilized in situations where the suspicion of alcohol abuse is high but the patient denies alcohol abuse. Carbohydrate-deficient transferrin (CDT) is elevated in alcohol abuse but is a less useful marker in females [21, 22] and in patients with cirrhosis [23]. Combinations of CDT, MCV, and/or GGT can help improve sensitivity and specificity [24–26] for heavy alcohol consumption.
Imaging
For patients with alcohol abuse who have significant fat in the liver (≥30 % fat), ultrasound will detect diffuse hyperechoic texture, with a sensitivity of 91 % and a specificity of 93 %. The sensitivity is only 64 % with fatty infiltration less than 30 % [27]. CT scan without contrast is highly predictive of fatty liver when the liver-to-spleen attenuation ratio is more than ten Hounsfield units [28]. MRI is the best tool but is more expensive, with a sensitivity of 95 % and specificity of 98 % [29]. When trying to identify advanced fibrosis or cirrhosis, ultrasound has lower sensitivity (50–70 %) with specificity of 88 % [30]. Multiphase CT scan and MRI are superior in identifying cirrhosis and its complications, including collateral circulation, vascular thrombosis, hepatocellular carcinoma, or other focal liver lesions. Ultrasound is the most commonly used initial imaging technique, helpful in identifying biliary dilation in a patient with jaundice and in the detection of ascites.
Liver Biopsy
In the presence of a history of alcohol abuse associated with typical liver enzyme elevations, diagnosis is very reliable with sensitivity of 91 % and specificity of 97 % [31], and liver biopsy is often not performed in the clinical (non-research) setting. Patients with atypical presentation or who have markers of other types of liver disease (autoimmune hepatitis, hemochromatosis, viral hepatitis, etc.) are good candidates for liver biopsy. Liver biopsy helps to clarify the diagnosis and will give the stage of disease, which is useful in deciding if surveillance for hepatocellular carcinoma is needed. Liver biopsy also differentiates simple steatosis from steatohepatitis but is rarely needed for this purpose. The histologic findings of alcoholic steatohepatitis include centrilobular steatosis, hepatocyte ballooning, Mallory bodies, perivenular fibrosis, pericellular fibrosis, and mixed inflammatory infiltration of neutrophils and lymphocytes. Megamitochondria are often seen in cases of recent alcohol abuse. Patients with cirrhosis frequently have micronodular changes and bile duct proliferation. Less often, patients have mixed macro- and micronodular cirrhosis.
Alcoholic Liver Disease Modifiers
Introduction
Many people drink heavily, yet only a limited number (~35 %) develop more advanced liver diseases (alcoholic hepatitis or cirrhosis). Thus, there must be modifying factors that either prevent or facilitate disease activity/progression. These modifiers can either be fixed (e.g., genetics) or can undergo intervention (e.g., smoking, diet). We review ten disease modifiers of particular importance to ALD (Table 8.2). Continued drinking, the most compelling modifier, is discussed throughout this chapter.
Table 8.2
Disease modifiers
Continued drinking |
Age, Sex |
Race |
Diet/Nutrition |
Genetics/Epigenetics/Family History |
Smoking |
Obesity |
Occupational/Environmental Exposure |
Medications/Drugs of Abuse |
Other Liver Diseases |
Gender and Age Differences
Males and females have differences in the absorption, distribution, and metabolism of alcohol [32, 33]. Females have a lower proportion of body water than males of equal body weight; therefore, they can achieve higher concentrations of blood alcohol even with equivalent amounts of alcohol [34]. Some investigations show that the gender variability in peak blood alcohol concentrations following equivalent low doses of alcohol could be due to the differences in first-pass metabolism of alcohol in the gastrointestinal tract, which was significantly correlated with gastric alcohol dehydrogenase (ADH) activity . Gastric ADH activity was lower in females [35, 36]. However, other studies have demonstrated no gender differences in the first-pass metabolism of alcohol [37] and indicated that first-pass metabolism was evident only with the ingestion of relatively low doses of ethanol and when gastric emptying is slow [38].
Epidemiological data also show that females are more susceptible to alcohol-related liver damage than men [39]. Women can develop ALD as alcoholic cirrhosis and alcoholic hepatitis at younger ages, at lower rates of daily alcohol intake, and at lower cumulative exposure to alcohol than the males [9]. Two hypotheses have been put forward to explain this gender-based difference. The first is related to gender-based variability in alcohol pharmacokinetics (PK), and the second is related to gender-based differences in the metabolic processes that are influenced by chronic alcohol use, such as the generation of free radicals, fatty acid metabolism, and endotoxins [40]. The mechanism for the former may be a larger liver volume or enhanced function in males, including the stimulation of alcohol-metabolizing enzymes in the liver, resulting in an alteration in the PK of alcohol. The latter is thought to be related, at least in part, to gender-based differences in portal endotoxemia, hepatic inflammatory responses, and the activation of Kupffer cells [41–43]. Both mechanisms may be related to fundamental differences between women and men in sex steroids, including estrogen, and their influence on alcohol metabolism as well as on liver disease susceptibility.
The role of age–gender interactions in response to the effects of alcohol is of interest, although there has been limited research in this area. The elderly are thought to be more sensitive to alcohol and show greater impairment than younger groups. However, it is not clear if these changes are due to pharmacokinetic or pharmacodynamic factors [44]. Pharmacokinetic changes, including a decrease in circulation volume, can result in increased alcohol levels and therefore increased impairment, in older participants following standard doses of alcohol. Age is a predictor for ALD prognosis [45]. Age and gender (and their potential interaction) also have been reported in various stages of ALD as risk factors that were associated with disease severity [46].
Race
Data are relatively consistent concerning race/ethnicity and ALD. Research from large multicenter Veterans Affairs Cooperative Studies showed that alcoholic cirrhosis was more frequent in Hispanics (73 %) than in non-Hispanic White/Caucasians (52 %) and African-Americans (44 %) with acute alcoholic hepatitis. African-Americans also were more likely to have hepatitis C as a confounder. Data from the Fourth National Health and Nutrition Examination Survey (NHANES IV) was used to evaluate ethnic differences and AST/ALT values. Mexican-American men and women were the most likely to have elevated aminotransferase activity. Among men, Mexican-Americans were more likely than whites to be heavy/binge drinkers. African-Americans have consistently shown to be more likely to have hepatitis B or hepatitis C [47]. Recent studies at the University of California Davis Medical Center showed that Hispanic patients presented 4–10 years earlier than their White/Caucasian counterparts in all states of severity of ALD [48]. There were more obese Hispanic patients than White/Caucasian patients.
Diet/Nutrition
Diet and nutrition play a major role in ALD, and patients with ALD show various degrees of nutritional deficiency [49]. Malnutrition is a multifactorial consequence of ALD [50] and has been best studied in patients with alcoholic hepatitis (AH) (which has a high association with malnutrition [51]). The most comprehensive reports on malnutrition in ALD have come from large studies conducted by the Veterans Health Administration (VA) Cooperative Studies Program in patients having AH [51–54]. Almost every AH patient showed some degree of malnutrition [52]. Further, approximately 50 % of patients’ energy intake came from alcohol. Although calorie intake was frequently not inadequate, intake of protein and critical micronutrients was often deficient. Importantly, the severity of liver disease and mortality correlated the severity of malnutrition (Fig. 8.1). Patients were given a balanced 2500-kcal hospital diet and encouraged to consume the diet. Voluntary oral food intake correlated in a stepwise fashion with 6-month mortality data. Thus, patients who voluntarily consumed more than 3000 kcal/day had virtually no mortality, whereas those consuming less than 1000 kcal/day had greater than 80 % 6-month mortality [51] (Fig. 8.1). Importantly, anorexia was a frequent finding in these AH patients, and it correlated with severity of liver disease. Moreover, alcohol consumption itself can also lower food craving [55].
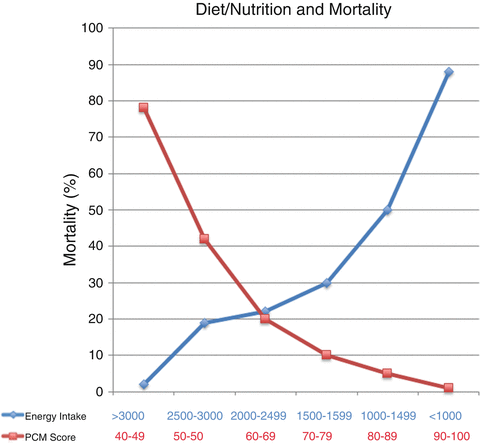
Fig. 8.1
Data from VA Cooperative Studies demonstrate a dose–response relationship between nutrition and outcome. As shown in red, the protein–calorie malnutrition (PCN score) decreased in an inverse, indirect relation with mortality. A normal score is 90–100, and the lower the score, the more severe the malnutrition. Similarly, voluntary calorie intake directly correlated with mortality (shown in blue). Those who ate ≤1000 cal had >90 % mortality, while those who ate >3000 cal had virtually no deaths
Dietary fat represents a macronutrient dietary modifier for ALD. Many investigations have shown that dietary saturated fat protects against alcohol-induced liver disease in preclinical models using rodents, whereas dietary unsaturated fat, enriched in linoleic acid (LA) in particular, promotes alcohol-induced liver damage [56–58]. LA is enzymatically converted to bioactive oxidation products, OXLAMs, primarily via the actions of 12/15-lipoxygenase (12/15-LO) or nonenzymatically via free radical-mediated oxidation in response to oxidative stress. LA is the most abundant polyunsaturated fatty acid in the human diet [59] and in human plasma and membrane lipids. Dietary intake of LA has more than tripled over the past century. We have shown that an LA-enriched diet enhances intestinal inflammation, endotoxemia, Toll-like receptor activation, and liver injury in a mouse model of ALD [60, 61]. We postulate that the type of dietary fat consumed may explain, at least in part, why only some people who drink heavily develop ALD.
Zinc deficiency is a micronutrient deficiency commonly observed in ALD. Zinc is an essential trace element required for normal cell growth, development, and differentiation. It is a critical component in many zinc proteins/enzymes, including critical zinc transcription factors. Some of the mechanisms for zinc deficiency/altered metabolism include decreased dietary intake, increased urinary excretion, and alterations in certain zinc transporters. Importantly, oxidative stress (through modification of the cysteines that retain zinc) may also cause loss of zinc from critical zinc finger proteins [62]. Zinc deficiency may manifest itself in many ways in liver disease, including skin lesions, poor wound healing, liver regeneration, altered mental status, or altered immune function. Zinc supplementation has been documented to block/attenuate experimental ALD through multiple processes, including stabilizing gut-barrier function, decreasing endotoxemia, decreasing proinflammatory cytokine production, decreasing oxidative stress, and attenuating apoptotic hepatocyte death [62, 63]. Clinical trials in human liver disease are limited in size and quality, but it is clear that zinc supplementation reverses clinical signs of zinc deficiency in patients with ALD (some studies suggest improvement in liver function in both ALD and hepatitis C following zinc supplementation). The dose of zinc used for treatment of liver disease is usually 50 mg of elemental zinc taken with a meal to decrease the potential side effect of nausea.
Smoking
Nicotine dependence is common among people with alcohol use disorders (AUD) , and the amount of smoking directly correlates with level of alcohol consumption and the severity of alcohol problems. Alcohol consumers, including heavy users or those with an AUD, are more likely to smoke cigarettes and be nicotine dependent, and the level of nicotine dependence/smoking correlates directly with levels of alcohol consumption [64]. Subjects with AUD are also more likely to have started smoking earlier in adolescence than smokers without an AUD [65, 66]. There may be common genetic factors in both smoking and alcohol use behaviors [67, 68].
Animal studies have established the role of nicotinic receptors in alcohol reward and provide strong evidence that alcohol and nicotine may act on the same brain pathways—particularly the mesolimbic dopamine system—to exert their rewarding effects and modulate consumption [69, 70]. Furthermore, nicotine administration has been found to increase alcohol consumption, particularly in men [71]. Nicotine is more reinforcing in alcohol-dependent people than in those who have never been dependent, and it increases alcohol consumption in male smokers who drink below hazardous levels.
Cigarette smoking can adversely impact certain hepatic functions and has been associated with severity in ALD in humans. In preclinical studies, cigarette smoking has been shown to have adverse effects on cytochrome P450 and UDP-glucuronosyltransferase activity in the liver [72]. Cigarette smoking can induce oxidative stress (which plays a mechanistic role in ALD). Further, nicotine-derived nitrosamine ketone (NNK) has been shown to have major role in the pathogenesis of steatohepatitis in a “chronic-plus-binge” rat model of alcoholic liver disease [73]. Cigarette smoking has also been shown to be a risk factor in the development of human alcoholic cirrhosis , especially in those smoking one or more packs per day [74]. Active tobacco use is a significant independent predictor of mortality (p = 0.03) [75].
BMI, Excess Weight, and Obesity
The obesity epidemic poses serious and multifaceted health problems in the USA, and recent data from 2011 to 2012 has shown that approximately 35 % of adults and 17 % of children and adolescents are affected by obesity [76]. Significant differences exist by ethnicity, age, and gender, with respect to the prevalence of overweight/obesity [77]. Excess weight and obesity have been known for decades to have negative consequences to liver health [78], and overweight/obesity has been considered as a risk factor for multiple diseases [79]. Obesity has been shown to have a close association with various forms and stages of ALD [46]. Overweight patients with no alcohol drinking history may have nonalcoholic fatty liver disease (NAFLD) with or without fibrosis [80], and/or cirrhosis, and, potentially, hepatocellular carcinoma [81]. Excessive fat in humans and elevated free fatty acids in patients with ALD could contribute to liver injury [82]. Body mass index (BMI) and fibrosis of the liver are positively correlated, and BMI is an independent risk factor for fibrosis in ALD [83]. The presence of overweight/obesity for at least 10 years was independently correlated with the presence of cirrhosis [46]. Excess body weight in patients with heavy alcohol consumption could markedly increase the severity of steatosis and is a risk factor for the development of more advanced stages of ALD, namely, acute alcoholic hepatitis and cirrhosis.
Genetics/Epigenetics/Family History
Recent studies have shown that both genetic and epigenetic factors are important for disease pathogenesis and progression in alcoholic liver disease (ALD) . The genetic variations are often associated with conformational changes in protein structures and functions due to single nucleotide polymorphisms (SNPs) . Genome-wide association studies (GWAS) have identified around 3.1 million SNPs that can contribute to disease states, and these SNPs may increase or decrease the function of encoded proteins. In ALD, polymorphisms of alcohol-metabolizing enzymes such as ADH and CYP2E1 as well as antioxidant enzymes and cytokine coding genes have shown strong correlation with the progression of ALD [84].
Epigenetic changes, including microRNAs, DNA methylation, and histone modifications, occurring in response to alcohol are known to produce diverse organ/tissue-specific effects. The role of miRNA is well recognized in ALD; some miRNAs play a causal role in disease development, whereas others may be mere associations. A decrease in miR-122 and induction in miR-155 expression has been reported in models of ALD; miR-122-deficient mice develop greater steatohepatitis and fibrosis, while TNF and CEBP are the targets of miR-155. Alcohol-induced alterations in miRNAs are associated with steatohepatitis [85] and fibrosis [86]. Chronic alcohol exposure also alters miRNAs that affect intestinal permeability during ALD [87]. For example, alcohol induces miR-212 which downregulates tight junction protein-1; also, miRNA-212 is higher in colon biopsies of patients with ALD. Additionally, other microRNAs such as miR-320, miR-486, miR-705, miR-1224, miR-27b, miR-214, miR-199a, miR-192, and miR-183 likely contribute to ALD [85].
DNA methylation of cytosine at C5 at CpG dinucleotide in promoter CpG islands silences transcription, whereas lack of methylation activates transcription. Studies in ALD show that the alcohol-induced decrease in s-adenosyl-l-methionine (SAM, the primary cellular methyl donor) can greatly influence DNA methylation and induce global as well as gene-specific changes leading to altered phenotype. Chronic alcohol consumption is reported to induce global DNA hypomethylation in the liver [88], whereas in peripheral blood cells, hypermethylation of DNA is observed after alcohol consumption in humans [89, 90]. In the context of hepatocellular carcinoma, aberrant DNA methylation was associated with alcohol intake and hypomethylation of the O6-methylguanine DNA methyltransferase gene [91].
Lastly, several in vivo and in vitro studies have established the important role of alcohol-induced histone modification in the development of ALD. Histone acetylation is regulated by the relative activities of histone acetyltransferase and histone deacetylase enzymes, which are both altered by alcohol. Reduced expression of SIRT 1, a class III HDAC, has been shown in alcohol-exposed hepatocytes and is known to regulate the lipid metabolism pathway. Our own studies support this notion. We have shown that dysregulation of hepatic HDAC expression plays a major role in the binge alcohol-induced hepatic steatosis and liver injury by affecting lipogenesis and fatty acid β-oxidation [92]. Site selective acetylation of histone H3 at lys 9 (H3AcK9), and not at H3 lys14, lys18, and lys23, has been observed in alcohol-exposed primary rat hepatocytes [93]. Along with histone acetylation, alcohol also alters histone methylation and phosphorylation in vitro in rat hepatocytes [94] and in vivo [95]. Further, different modifications at different sites in the same histone (e.g., lys-4, lys-9, ser-10, ser-28, H3) may occur on nucleosomes located in different chromatin domains [96].
An interplay that exists between the various epigenetic mechanisms can determine downstream chromatin remodeling and gene expression; for example, DNA hypermethylation can trigger histone deacetylation, and lower histone acetylation increases DNA methylation. Detailed studies are needed to better understand the cross talk and hierarchical order in epigenetic mechanisms in ALD and how interventions may positively modify epigenetics.
Occupational/Environmental Exposure
Exposure to potential toxins in either the workplace or environment can cause hepatotoxicity which can be exacerbated by alcohol. Vinyl chloride (VC) represents a potential industrial/workplace exposure whose toxicity may be exacerbated by alcohol. We reported that VC induced histologic steatohepatitis that was indistinguishable from alcohol-induced steatohepatitis, and we termed this toxicant-associated steatohepatitis (TASH) [97]. VC is metabolized in a strikingly similar fashion to ethanol, and this could potentially account for the observed similarities between TASH and AH. Although initial studies suggested that, at low substrate concentrations (below 100 ppm), VC is metabolized by a pathway involving alcohol dehydrogenase, most studies have concentrated on the role of CYP2E1 as the initial catalyst of VC metabolism. At concentrations up to ≈220 ppm, VC is metabolized by CYP2E1, forming the highly reactive genotoxic epoxide, chloroethylene oxide. Chloroethylene oxide either spontaneously or enzymatically is converted to chloroacetaldehyde. In either pathway, chloroacetaldehyde is formed. Thus, both alcohol and VC are metabolized through similar pathways to a toxic aldehyde metabolite. Our preliminary research in experimental animals suggests that co-exposure may be more toxic than either agent alone.
Arsenic represents an environment for exposure that has great potential to exacerbate alcohol-induced liver injury. Alcohol promotes arsenic absorption. Both arsenic and alcohol increase reactive oxygen species, deplete hepatic GSH levels, impair mitochondrial function, and cause alterations in DNA methylation [98]. With environmental exposures, there are usually multiple contaminants rather than just one compound such as arsenic. A recent Canadian study evaluated a cocktail of 22 contaminants thought to be clinically relevant (northern contaminant mixture) and showed that both a high-fat diet and alcohol increased fatty liver and liver injury in exposed mice [99].
In summary, because of similarities in metabolism and mechanisms of liver injury, occupational/environmental exposures will be highly relevant to the development/progression of ALD, and these exposures are often overlooked.
Medications/Drugs of Abuse
Alcohol and other drugs (including prescription medications, over-the-counter agents, and illicit drugs) may interact to cause hepatotoxicity, and this hepatotoxicity sometimes can be misdiagnosed as traditional ALD. Acetaminophen hepatotoxicity is a classic form of liver injury that can be enhanced by chronic alcohol ingestion. Chronic alcoholics can be more susceptible to acetaminophen hepatotoxicity for a variety of reasons. They frequently eat diets containing inadequate protein which may adversely affect hepatic glutathione stores. Alcoholics may develop gastrointestinal problems, such as gastritis or pancreatitis, with nausea, vomiting, and decreased food intake that may decrease hepatic glutathione stores. Fasting also has been shown to induce hepatic drug metabolism (P450) in rats, and ethanol and certain higher chain alcohols are well-documented inducers of cytochrome P450 (P450-2E1). Lastly, chronic alcoholics are frequently deficient in nutrient antioxidants, such as selenium, vitamin E, and zinc, and this may augment oxidant liver injury. Thus, for multiple reasons, chronic alcoholics may be predisposed to acetaminophen liver injury [100]. This form of liver injury clinically presents with very elevated AST and ALT levels (often >1000 IU/mL) that distinguish it from ALD.
Alcohol abuse is common among HIV-infected patients. Alcohol abuse frequently lessens compliance with antiretroviral therapy and can also enhance hepatotoxicity of certain HAART regimens [101]. Alcohol may also interact with illicit drugs such as 3,4-methylenedioxymethamphetamine (MDMA; ecstasy) which is an amphetamine-derivative drug commonly consumed at rave parties along with other drugs including alcohol [102]. Alcohol has been shown to enhance MDMA hepatotoxicity in mice.
Other Liver Diseases
Alcohol abuse may accelerate other liver diseases and other liver disease may accelerate ALD [103]. Hepatitis C is much more prevalent (up to tenfold higher) in alcoholics than in the general population and is greatest in those with more advanced liver disease. Alcohol and hepatitis C are thought to interact to cause accelerated liver disease through multiple different pathways including increased oxidative stress, enhanced viral replication, hepatocyte apoptosis, altered gut-barrier function, immune dysfunction, and alteration in epigenetics (miR-122), to name only a few [104].
Distinguishing between patients with alcoholic liver disease and those with secondary iron overload from hereditary hemochromatosis can sometimes be difficult. Patients with alcoholic cirrhosis can have elevated serum iron and ferritin levels and increased hepatic iron levels suggestive of hereditary hemochromatosis [105]. Moreover, 15–40 % of patients with hereditary hemochromatosis consume more than 80 g of alcohol daily [106]. Performing genetic testing for hereditary hemochromatosis will assist in making the correct diagnosis. There also is the possibility that being a heterozygote for hemochromatosis or some other hereditary diseases, such as α1 antitrypsin deficiency, may accelerate the course of ALD.
Natural History of Alcoholic Liver Disease
The phenotypical manifestation of alcohol consumption in the liver, and general health status of an individual, is determined by a complex interplay of varying elements such as drinking habits (duration and quantity of alcohol consumption), environmental agents, and individual factors, as discussed previously [107]. Chronic alcohol consumption can lead to a spectrum of liver injuries which can occur sequentially, separately, or simultaneously in the same patient [108].
Alcoholic steatosis is the most common and initial manifestation of alcoholic liver disease. This lesion is pathologically characterized by both microvesicular and macrovesicular fat accumulation within the hepatocytes, with minimal inflammatory response or hepatic fibrosis [109]. Patients with steatosis are usually asymptomatic and are often diagnosed incidentally on abdominal imaging (usually in the form of transabdominal ultrasound or computed tomography (CT) scan). Patients typically have normal to very mild elevations of their liver enzymes, such as gamma-glutamyl transpeptidase (GGT), alanine aminotransferase (ALT), and aspartate aminotransferase (AST). Serum bilirubin and other markers of hepatic function (international normalized ratio (INR), albumin levels) also tend to be normal. Alcoholic steatosis is a relatively benign condition which is reversible with abstinence; however, it has the potential to progress to alcoholic hepatitis or cirrhosis in an accelerated fashion in a minority of patients who continue to drink heavily [110].
Early studies by Dr. Charles Lieber in volunteers demonstrated the relative ease with which alcohol consumption causes fatty liver [111, 112]. In the first study, five volunteer subjects with a history of alcoholism were fed alcohol in a clinical research setting. All subjects had previously abstained from alcohol for 2–5 months. The study lasted 18 days. Patients were given 25 % of calories as protein (high), 25 % of calories as fat (low), and 50 % as carbohydrates. Alcohol was substituted for carbohydrates and was slowly increased so that after 8 days on the study, subjects were receiving 46 % of total calories as alcohol. Some subjects had interval liver biopsies during this study, and two had liver biopsies performed 1 month after alcohol was discontinued. Alcohol consumption caused histologic and biochemical fatty liver and ultrastructural changes in the liver. This occurred in spite of the fact that subjects received a nutritious diet. This highlighted the fact that alcohol itself is hepatotoxic in spite of a nutritious diet. In a second study, healthy volunteers who were never drinkers were given alcohol anywhere from 2 days to 2 weeks. Four different regimens of alcohol feeding were administered. One group received alcohol for only 2 days in addition to a standard diet, and another group received alcohol for 2 days in addition to a high-protein/low-fat diet. Importantly, both groups developed hepatic steatosis. Longer duration of feeding involved isocaloric substituting of alcohol for carbohydrates. Again, all subjects developed fatty liver on liver biopsy. These results clearly demonstrate that normal nonalcoholic subjects who regularly consume alcohol even for a relatively short period of time can develop fatty liver, and histologic changes were independent of nutritional factors.
The paramount histologic finding determining the natural history of alcoholic liver disease is alcoholic hepatitis. This is a necroinflammatory process characterized by predominant neutrophilic infiltration, ballooning degeneration of hepatocytes, and hepatocyte necrosis [113]. The development of this key clinical and histologic entity represents a “fork in the road” in relation to the natural history of ALD, with important short-term and long-term implications. Acutely, the development of alcoholic hepatitis is associated with a significant increase in proximal mortality and the potential to develop portal hypertension and its complications, even without the development of major fibrosis [114]. In the long term, for those who survive, this disease process is associated with an accelerated course of fibrosis progressing to cirrhosis in 40 % of cases [115]. These distinctions in the natural progression of alcoholic liver disease also have therapeutic implications, explaining why a subset of alcoholics with inflammatory features are candidates for treatment with anti-inflammatory agents (i.e., corticosteroids and pentoxifylline) in an attempt to reduce proximal mortality, whereas patients without these inflammatory features may be better candidates for treatment geared toward reducing long-term hepatic injury/cell death and hepatic dysfunction or possibly enhancing liver regeneration.
Due to the important acute prognostic implication of alcoholic hepatitis , multiple scoring systems have been developed to assess the severity of liver disease in terms of patient survival in order to stratify patients to proven treatment modalities. The Child–Turcotte–Pugh (CTP) , the oldest scoring system, incorporates the serum bilirubin level, albumin level, PT, and the severity of ascites and hepatic encephalopathy in assigning a numerical score that is used to categorize patients (class A = scores 1–6, class B = scores 7–9, class C = scores 10–15), with a higher score denoting more severe disease [116]. This scoring system has fallen out of favor in grading alcoholic hepatitis due to subjectivity in grading among other things. Since 2002, the United Network for Organ Sharing (UNOS) has utilized the MELD score to grade the severity of liver disease in patients awaiting liver transplant. This represents a more objective analysis, utilizing only the serum bilirubin, creatinine, and INR [117]. The use of this scoring system is a reflection of the reason for which this scoring system was initially intended to assess—the short-term prognosis of cirrhotic patients undergoing transjugular intrahepatic portosystemic shunt (TIPS) procedures [118]. The use of this scoring system has been validated in multiple studies to accurately predict the 3-month mortality of patients with liver disease, especially those patients awaiting liver transplantation [119].
Related posts:
 Nonalcoholic Fatty Liver Disease in Children
Diagnostic Considerations and Clinical End Points for Nonalcoholic Steatohepatitis
Hepatic and Extrahepatic Malignancies in NAFLD
Nonalcoholic Fatty Liver Disease in Children
Diagnostic Considerations and Clinical End Points for Nonalcoholic Steatohepatitis
Hepatic and Extrahepatic Malignancies in NAFLD
 Hepatic and Extrahepatic Malignancies in Alcoholic Liver Disease
Hepatic and Extrahepatic Malignancies in Alcoholic Liver Disease
 Animal Models of Alcoholic Liver Disease
Animal Models of Alcoholic Liver Disease
 Epidemiology and Risk Factors for Alcoholic Liver Disease
Epidemiology and Risk Factors for Alcoholic Liver Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


