CHAPTER 104 Celiac Disease and Refractory Celiac Disease
DEFINITIONS
Celiac disease is characterized by small intestinal malabsorption of nutrients after the ingestion of wheat gluten or related proteins from rye and barley, villus atrophy of the small intestinal mucosa, prompt clinical and histologic improvement following strict adherence to a gluten-free diet, and clinical and histologic relapse when gluten is reintroduced.1 The many other names used to identify patients with this condition, including nontropical sprue, celiac syndrome, adult celiac disease, idiopathic steatorrhea, and primary malabsorption, among others, are testimony to the confusion of the past. The term celiac disease is recognized widely and is used in this chapter; celiac sprue and gluten-sensitive enteropathy are acceptable alternative terms.
Celiac disease exhibits a spectrum of clinical presentations (Fig. 104-1). Atypical celiac disease is fully expressed gluten-sensitive enteropathy manifest only by extraintestinal symptoms and signs including short stature, anemia, osteoporosis, and infertility. Silent celiac disease is fully expressed gluten-sensitive enteropathy usually found after serologic screening in asymptomatic patients. The atypical and silent variants are more common than classic or typical celiac disease, which is fully expressed gluten-sensitive enteropathy found in association with the classic gastrointestinal symptoms of malabsorption.
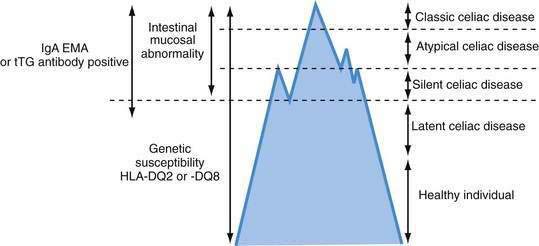
A combination of serologic, genetic, and histologic data also has led to the identification of two other types of celiac disease. Patients with latent celiac disease have normal villus architecture on a gluten-containing diet but, at another time, have had or will have gluten-sensitive villus atrophy. For example, a patient who had celiac disease in childhood and recovered completely on a gluten-free diet might have latent celiac disease later in life on resumption of a normal diet. Patients with potential celiac disease have never had a biopsy consistent with celiac disease but show immunologic abnormalities characteristic for the disease, such as a positive immunoglobulin (Ig)A antibody to endomysium (or tissue transglutaminase [tTG]) or increased intraepithelial lymphocytes (IELs) in the small intestine. These patients often have a genetic predisposition to celiac disease, especially human leukocyte antigen class II DQ (HLA-DQ2), an affected first-degree relative, or both. The probability of their eventually developing celiac disease is unpredictable.2
Refractory celiac disease, also known as unclassified or intractable celiac sprue, is defined as symptomatic, severe small intestinal villus atrophy that mimics celiac disease but does not respond to at least six months of a strict gluten-free diet. This is a diagnosis of exclusion that is not accounted for by inadvertent gluten ingestion, other causes of villus atrophy, or overt intestinal lymphoma.1,3
HISTORY OF CELIAC DISEASE
Celiac disease was recognized as a clinical entity by Aretaeus the Cappadocian in the first century ad.4 The name sprue was coined in the 18th century and is derived from the Dutch word spruw, which means “aphthous disease,” so named because of the high prevalence of aphthous mouth ulcers in these patients. In 1888, Samuel Gee published his paper “On the Coeliac Affection,” which described many of the clinical features of celiac disease in patients of all age groups and concluded, “If the patient can be cured at all it must be by means of the diet.”5 It was not until the middle of the 20th century, however, that the link between certain cereals and celiac disease was made by Willem Karel Dicke, a Dutch pediatrician. He became convinced that the consumption of bread and wheat flour was directly responsible for the deterioration in patients suffering from this condition.6 During World War II, cereals used to make bread were particularly scarce in the Netherlands, and during this time, children with celiac disease improved, only to relapse after the supply of cereal was re-established at the end of the war. It was this serendipitous observation that led to the finding that wheat ingestion exacerbated celiac disease. Subsequent work by van de Kamer and coworkers showed that it was the water-insoluble portion, or gluten moiety, of wheat that produced intestinal injury in patients with celiac disease.7
In 1954, Paulley provided the first accurate description of the characteristic intestinal lesion in patients with celiac disease.8 With the development of effective peroral suction biopsy instruments in the late 1950s, Rubin and coworkers demonstrated that celiac disease in children and idiopathic or nontropical sprue in adults were identical diseases with the same clinical and pathologic features.9
Since the 1980s, we have seen substantial advances in our understanding of the genetic, immune, and molecular mechanisms fundamental to the pathogenesis of celiac disease. In 1986, Howell and associates observed that celiac disease was associated with specific HLA-DQ2 haplotypes.10 In 1993, Lundin and colleagues demonstrated that the DQ2 gene products preferentially present gluten-derived gliadin peptides to intestinal mucosal T cells in celiac patients.11 Subsequently, the enzyme tTG (more specifically tTG type 2 [tTG-2]) was identified as a celiac autoantigen, leading to more accurate serologic diagnostic tests.12
In 1998, Molberg and colleagues reported that modification of gliadin by host tTG enhances gliadin-specific celiac disease T-cell responses.13 The identification of specific tTG-modified deamidated gliadin peptides (DGPs) as dominant α-gliadin T-cell epitopes has highlighted the pivotal role played by tTG in the pathogenesis of celiac disease.14 This discovery already has led to more accurate antigliadin antibody serologic testing using DGPs as capture antigens, and it might pave the way for antigen-specific immunotherapy.
The key role played by IELs in the development of refractory celiac disease and enteropathy-associated T cell lymphoma (EATL) continues to evolve.15 Studies also point to the importance of interleukin (IL)-15, a potent proinflammatory cytokine at the interface between innate and adaptive immunity in the pathogenesis of both celiac disease and refractory celiac disease.16
Epidemiologic studies using endomysial antibody (EMA) and tTG serology have substantially increased estimates of celiac disease prevalence in the United States and elsewhere.17 This in turn has led to renewed interest in potential nondietary treatments including glutenases, modifiers of tight junction function, tTG inhibitors and immune-based interventions, bringing celiac disease therapy into a new era.18,19
EPIDEMIOLOGY
The term celiac iceberg was coined to describe the wide variations in the nature and intensity of clinical presentation of which overt celiac disease is only the emerging peak (see Fig. 104-1). The discovery of the large immersed part of the celiac iceberg has transformed the status of celiac disease, long considered a rare disease, particularly in adults, to that of a common health problem. Because we are uncertain of the depth and breadth of the celiac iceberg, the true prevalence of celiac disease remains unknown.
Serologic testing has demonstrated that silent celiac disease, characterized by positive serology and villus atrophy with few or no symptoms, is approximately seven times more common than symptomatic celiac disease.20 A Finnish study of 3654 schoolchildren of ages 7 to 16 years, using two serologic screens with antiendomysial and tTG antibodies, demonstrated the heterogeneity of the celiac iceberg, with one of every 99 children having biopsy-proved celiac disease.21 Only 10 of 56 subjects with a positive serology had overt symptoms of celiac disease. Two subjects with positive antibodies and at risk for celiac disease because of HLA-DQ2 haplotype had normal mucosa, but both had increased epithelial expression of HLA-DR suggesting mild intestinal inflammation, and one had high counts of IELs; these patients might represent cases of potential disease susceptible to evolving into overt celiac disease. Five patients who had HLA-DQ2 and positive antibodies when studied in 1994 had negative antibodies on repeat testing in 2001; their intestinal biopsies were normal, but all had increased HLA-DR expression, and four of the five had markedly increased numbers of IELs. This latter finding might indicate a variation in the natural history of celiac disease, occasionally seen in teenagers, in whom gluten sensitivity fluctuates with time.
Celiac disease shows a marked geographic variation, with the highest incidence in Western Europe. The condition is more common in Scandinavian and Celtic populations, where the prevalence has been reported to be as high as 1 in 9921 and 1 in 122,22 respectively. The prevalence is similarly high in Italy20 and the southeastern region of Austria.23 The prevalence in Denmark is 40-fold lower than that in Sweden,24 suggesting considerable variation in prevalence among geographically proximate populations. Factors such as predominant HLA haplotype, timing of introduction of gluten into the diet, differences in the gliadin concentration of infant formulas, and interobserver variation in interpreting small intestinal biopsy findings might explain the differences in prevalence.25 Celiac disease also is found in areas to which Europeans have emigrated, notably North America, South America, and Australia.
Epidemiologic studies in the United States, where the disease only recently has attracted much attention, underscore the varying clinical presentation of celiac disease and indicate that the prevalence in the United States is comparable with that in Western Europe. A large multicenter study by Fasano and coworkers17 determined the prevalence of antiendomysial antibodies in more than 13,000 at-risk and not-at-risk American subjects and found the prevalence of antiendomysial antibodies to be 1 in 22 and 1 in 39 among first-degree and second-degree relatives of subjects with celiac disease, respectively.17 A prevalence of 1 in 56 was documented among patients with celiac-like gastrointestinal symptoms or with associated disorders. Of most significance, these investigators found a prevalence of antiendomysial antibodies of 1 : 133 among 4126 “not-at-risk” subjects.
Although celiac disease is rare in the predominantly rice-eating area of southern India, it is prevalent in the Bengal and Punjab provinces of northwest India, where wheat rather than rice has, for many generations, has been a staple of the diet. The condition has been reported in blacks, Arabs, Hispanics, Israeli Jews, Sudanese of mixed Arab-black descent, and Cantonese and is particularly high among the Saharawi population in northwest Africa.26 The condition rarely affects people of purely sub-Saharan African, African-Caribbean, Chinese, or Japanese descent. Some authors have noted a female-to-male ratio of 2 : 1, whereas others have reported ratios as low as 1.3 : 1 but still suggesting a female predominance.
PATHOLOGY
Celiac disease affects the mucosa of the small intestine; the submucosa, muscularis propria, and serosa usually are not involved. The mucosal lesion of the small intestine in celiac disease can vary considerably in severity and extent.9 This spectrum of pathologic involvement might contribute to the striking variations in the clinical manifestations of the disease.
Examination under magnification of the small intestinal mucosal surface in severe untreated celiac disease reveals a flat mucosal surface with complete absence of normal intestinal villi. Histologic examination of tissue sections confirms this loss of normal villus structure (Fig. 104-2A). The intestinal crypts are markedly elongated and open onto a flat absorptive surface. The total thickness of the mucosa is reduced only slightly in most cases, because crypt hyperplasia compensates for the absence or shortening of the villi. These architectural changes decrease the amount of epithelial surface available for digestion and absorption.9
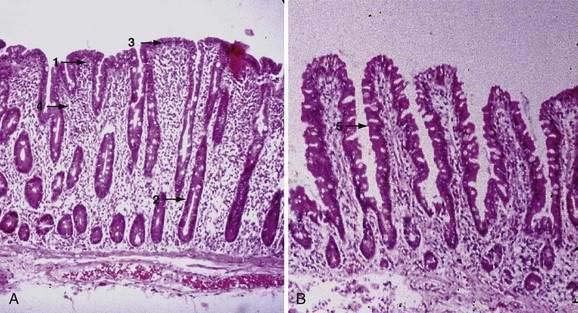
Structural abnormalities of tight junctions between damaged absorptive cells provide a morphologic explanation for the increased permeability of the mucosal barrier in celiac disease.27 The endoplasmic reticulum is sparse, reflecting the low level of synthesis of digestive enzymes, including disaccharidases and peptidases. Thus, mature absorptive cells are reduced in number and functionally compromised.
Unlike the absorptive cells, the undifferentiated crypt cells are markedly increased in number in patients with severe untreated celiac disease, and the crypts are therefore lengthened. Moreover, the number of mitoses in crypts is strikingly increased. Cytologic features and histochemistry of the crypt cells are normal by both light and electron microscopy. Studies of epithelial cell kinetics in untreated celiac disease suggest that “villus atrophy” is a misnomer because there is evidence for an actual increase in enteropoiesis in the crypts. Wright and colleagues28 estimated that intestinal mucosa from patients with celiac disease produces six times as many cells per hour per crypt as does normal small intestine and that the cell cycle time is halved, reflecting premature shedding. The experimental evidence suggests, therefore, that the central mechanism of villus shortening in celiac disease is a gliadin-associated toxic effect on maturing enterocytes that results in their premature loss into the intestinal lumen and a compensatory increase in enterocyte replication in the crypts. Such a mechanism would explain many of the histologic abnormalities described earlier.
The cellularity of the lamina propria is increased in the involved small intestine. The cellular infiltrate consists largely of plasma cells and lymphocytes. The number of IgA-, IgM-, and IgG-producing cells is increased two-fold to six-fold, but, as in normal mucosa, IgA-producing cells predominate.29 Polymorphonuclear leukocytes, eosinophils, and mast cells also can contribute substantially to the increased cellularity of the lamina propria. The number of IELs per unit area of absorptive epithelium (often reported as number of IELs per 100 enterocytes) is increased in untreated celiac disease.9 In the normal small intestinal mucosa, lamina propria T cells are predominantly CD4+ (helper/inducer) cells, whereas the IELs are mainly CD8+ (cytotoxic/suppressor) cells. In untreated celiac disease, this distribution of lamina propria T cells is maintained, but the density of cells in both compartments is increased.
Marsh30 pioneered the theory of a sequence of progression of the celiac lesion in the small intestinal mucosa. Starting with a normal, preinfiltrative (stage 0) mucosa, the initial observed event is an increase in IELs, followed by infiltration of the lamina propria with lymphocytes (stage 1). Crypt hyperplasia (stage 2) precedes villus atrophy (stage 3) and is observed only in the presence of lamina propria lymphocytosis, suggesting that IELs are not sufficient to induce intestinal architectural changes in celiac disease. Finally, total mucosal atrophy (stage 4) develops and is characterized by complete loss of villi, enhanced apoptosis, and crypt hyperplasia.
In untreated patients, the length of small intestinal involvement by the celiac disease lesion varies among individual patients and correlates with the severity of clinical symptoms. Thus, the patient with a severe lesion that involves the full length of the small intestine has more severe malabsorption than the patient with a severe duodenal lesion, a milder jejunal lesion, and a normal ileum. When the intestinal lesion does not involve the entire length of small bowel, the proximal intestine is usually the most severely involved; sparing of proximal intestine with involvement of the distal small intestine can occur, but it is rare. In some untreated patients with clinically mild celiac disease, even the proximal intestine shows only mild partial villus atrophy.9 It is important to note that an increase in IEL count alone is not sufficient to support the histologic diagnosis of celiac disease. This finding is nonspecific and is seen in many other conditions including bacterial overgrowth, mild peptic duodenitis, H. pylori infection, and in other autoimmune disorders. Thus, some shortening of the villi, crypt hyperplasia, cytologically abnormal surface cells, and increased lamina propria cellularity must be present to establish the diagnosis firmly.
Treatment with a gluten-free diet results in significant improvement in intestinal structure (see Fig. 104-2B). The cytologic appearance of the surface absorptive cells improves first, often within a few days. Tall, columnar absorptive cells with basal nuclei and well-developed brush borders replace the abnormal, immature cuboidal surface cells; the ratio of IELs to absorptive cells decreases. Subsequently, villus architecture reverts toward normal, with lengthening of the villi and shortening of the crypts; the lamina propria decreases in cellularity. The mucosa of the distal small intestine improves more rapidly than that of the more severely involved proximal bowel.30,31 In some patients, months or even years of gluten withdrawal may be required before the mucosa reverts to normal; indeed, some residual abnormality, which may be striking or subtle, often persists, possibly because of inadvertent gluten ingestion.32 In the debilitated patient with severe untreated celiac disease and associated nutritional deficiency states, pathologic changes may be present in many other organ systems besides the digestive tract. Finally, the mucosal lesion of celiac disease can be identical histologically to the mucosal response to injury typical of a wide range of other enteropathies (see “Differential Diagnosis”).
PATHOGENESIS
ENVIRONMENTAL FACTORS
Celiac disease is a model for autoimmune diseases with a defined environmental trigger. Early work involving physiologic digestion with pepsin and trypsin, followed by separation according to solubility properties, identified several wheat proteins as being responsible for the grain’s toxicity in celiac disease. Wheat protein exists in a number of storage forms that can be categorized into four general groups based on their solubility characteristics: prolamins (soluble in ethanol), glutenins (partially soluble in dilute acid or alkali solutions), globulins (soluble in 10% NaCl), and minor albumins (soluble in water). The term gluten encompasses both the prolamins and the glutenins. Although most toxicity studies have been performed with prolamins, there are data to suggest that glutenins also can damage the celiac intestinal mucosa.33
The prolamins of wheat are referred to as gliadins. Prolamins from other cereals also are considered to be gluten and are named according to their source (secalins from rye, hordeins from barley, avenins from oats, and zeins from corn). The taxonomic relationships of the major cereal grain families provide a framework on which their toxicities in celiac disease can be predicted (Fig. 104-3).34 Wheat, rye, and barley belong to the tribe known as Triticeae, and oats belong to a neighboring tribe known as Aveneae. Avenin is genetically less similar to gliadin than gliadin is to secalin and hordein. Despite their genetic differences, however, prolamins from oats, barley, wheat, and rye still have immunologic cross-reactivity because of their common ancestry.35 Grains that do not activate disease (rice, corn, sorghum, and millet) are separated still further from wheat, rye, and barley in terms of their derivation from the primitive grasses.
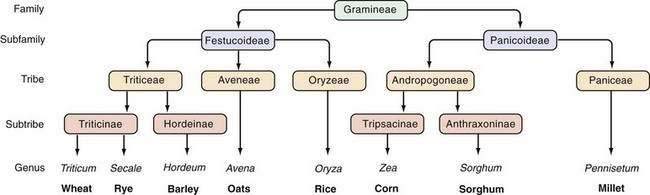
Figure 104-3. Taxonomic relationships of the major cereal grains.
(From Kasarda DD, Okita TW, Bernardin JE, et al. Nucleic acid [cDNA] and amino acid sequences of α-type gliadin from wheat [Triticum aestivum]. Proc Natl Acad Sci U S A 1984; 81:4712.)
Gliadin can be separated electrophoretically into four major fractions that range in molecular weight from 20 to 75 kd and exist as single polypeptide chains. These have been designated α-, β-, γ-, and ω-gliadins, and all four fractions appear to be toxic to patients with celiac disease.36 The complete amino acid sequences of several of the gliadins and related prolamins in grains other than wheat are known.33 In 2000, Anderson and colleagues14 identified a partially deamidated peptide, consisting of amino acids 56 to 75 of α-gliadin as a dominant epitope, responsible for activation of T cells in celiac disease. The complexity and diversity of the gliadin-specific T-cell response, however, is far greater than was previously appreciated, and persons with celiac disease can respond to a diverse repertoire of gluten peptides.37 Furthermore, the release of intracellular tTG leads to the deamidation of gluten proteins and an enhancement of T-cell responses to the resulting DGPs.14
In organ cultures, a synthetic peptide corresponding to amino acids 31 to 49 of α-gliadin has been shown to be toxic to intestinal mucosa and to induce epithelial lesions via recruitment of IELs. Peptide 31-49 does not activate intestinal CD4+ T cells from patients with celiac disease in vitro, but a related peptide corresponding to amino acids 31 to 43 is capable of activating peripheral CD4+ T cells isolated from patients with celiac disease and of inducing epithelial cell apoptosis and activating macrophages, thereby indicating a likely role for innate immune responses in disease pathogenesis.38 Gianfrani and colleagues39 reported that the α-gliadin-derived peptide corresponding to amino acids 123 to 132 is recognized by CD8+ T lymphocytes from patients with celiac disease and is associated with cytotoxic activity. By contrast, another peptide corresponding to amino acids 57 to 68 appears to function in adaptive immunity via stimulation of intestinal T cells in vivo but does not appear to be directly toxic to the intestinal mucosa of patients in vitro.37
It also is possible that immunologic similarities between gliadin protein motifs and enteric pathogens may be involved in the pathogenesis of an immunologic response to gluten antigens. This hypothesis was supported by a study in which analysis of α-gliadin demonstrated an amino acid region that was homologous to the 54-kd E1b protein coat of adenovirus 12, suggesting that exposure to the virus in a susceptible person could be involved in pathogenesis of celiac disease.40 Although patients with celiac disease have been reported to have a significantly higher prevalence of past adenovirus 12 infection than do control subjects,41 the role of adenovirus molecular mimicry in the pathogenesis of celiac disease has not been confirmed.
The reason oats are tolerated by almost all patients with celiac disease is not obvious, because the prolamin fraction of oats contains the same amino acid sequences (QQQPF, where Q = glutamine, P = proline, and F = phenylalanine) that in wheat gliadin have been shown to be toxic.42 A possible explanation for this paradox is that oats contain a relatively smaller proportion of this toxic prolamin moiety than do toxic gluten-containing cereals. Although a feature common to prolamins of wheat, rye, and barley is a high content of glutamine (∼30%) and proline (∼15%), the prolamins of oats have an intermediate content of these amino acids, and the nontoxic prolamins of rice, corn, and millet have an even lower content of them.43 This hypothesis is supported by collectively considering the studies on oat challenge in patients with celiac disease; these studies suggest that tolerance to oats might depend at least in part on the total amount consumed.44 Daily oats consumption of less than 40 to 60 g/day by patients whose celiac disease is in remission appears to be well tolerated.
The data on oats also highlight the important relationship between the amount of gluten consumed and the severity of disease manifestation. A 5- to 10-fold higher incidence of overt celiac disease in children from Sweden compared with Denmark (two populations with similar genetic backgrounds) has long been cited as evidence of the importance of environmental over genetic factors in pathogenesis of celiac disease. Subsequent studies found as much as a 40-fold difference in the gliadin concentration of Swedish compared with Danish infant formula.25 This finding suggests that early exposure of the immature immune system to significant amounts of gliadin is a prominent cofactor for the development of overt celiac disease, possibly by skewing the intestinal immune response to gliadin toward a T-helper 1 (Th1) T-cell response.
The age at which gluten is first introduced into an infant’s diet might also play a pivotal role in facilitating gluten tolerance or intolerance. In one study, early exposure to dietary gluten (within three months of birth) was associated with a five-fold increased risk for celiac disease compared with later gluten introduction (four to six months).45 In the same study, delaying gluten introduction (after 7 months of age) also was associated with a slightly increased risk for subsequent celiac disease (1.9-fold compared with the nadir at introduction at four to six months).
GENETIC FACTORS
Family studies that demonstrate frequent intrafamilial occurrence of celiac disease reflect the importance of genetic factors in its pathogenesis.44 Concordance for celiac disease in first-degree relatives ranges between 8% and 18% and reaches 70% in monozygotic twins.46 Our understanding of the nature of this genetic predisposition began with the significant observation by Howell and coworkers10 that celiac disease was associated with specific HLA-DQ2 haplotypes. HLA class II molecules are glycosylated transmembrane heterodimers (α and β chains) that are organized into three related subregions—DQ, DR, and DP—and encoded within the HLA class II region of the major histocompatibility complex on chromosome 6p. An important link to a genetic predisposition was provided by the isolation of gliadin-specific HLA-DQ2-restricted T-cell clones from celiac disease mucosa.11,47
It is now known that after gluten is absorbed, lamina propria antigen-presenting cells (probably dendritic cells) that express HLA-DQ2 or HLA-DQ8, present gliadin peptides on their α/β heterodimer antigen-presenting grooves to sensitized T lymphocytes expressing the α/β T cell receptor (TCR). These lymphocytes then activate B lymphocytes to generate immunoglobulins and other T lymphocytes to secrete cytokines, including interferon (IFN)-γ, as well as IL-4, IL-5, IL-6, IL-10, tumor necrosis factor (TNF)-α, and transforming growth factor (TGF)-β.48 These cytokines induce not only enterocyte injury but also expression of aberrant HLA class II cell-surface antigens on the luminal surface of enterocytes, possibly facilitating additional direct antigen presentation by these cells to the sensitized lymphocytes (Fig. 104-4).
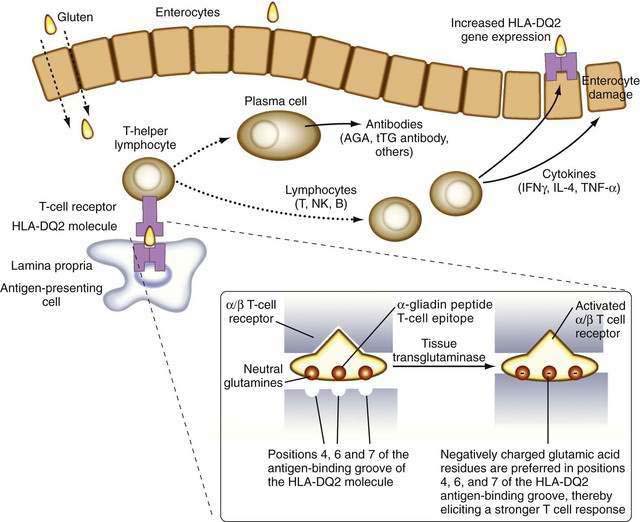
Only a minority of persons who express DQ2 actually develop celiac disease. HLA-DQ2 is expressed by approximately 35% of Europeans and their descendants, but it is rare in other populations (e.g., in sub-Saharan Africa or far eastern Asia). Thus, much of the genetic predisposition to celiac disease is conferred by genes other than those encoding HLA DQ molecules. The search for other genes that confer susceptibility to celiac disease has revealed numerous loci of interest on several different chromosomes, some of which also are associated with susceptibility to type 1 diabetes.49–52
IMMUNE FACTORS
There is substantial evidence implicating both humoral- and cell-mediated immune responses to gliadin and related prolamins in the pathogenesis of celiac disease. There is a two- to six-fold increase in the numbers of immunoglobulin-producing B cells in the lamina propria of the small intestine in untreated celiac disease patients.28 In addition, IgA and IgG serum antibodies to purified gliadin, all major fractions of gliadin, and DGPs can be detected in the sera of most patients with untreated celiac disease.14,53–56 Antigliadin antibodies (AGAs), however, do not appear to be essential for the pathogenesis of celiac disease and might simply reflect a nonspecific response to the passage of incompletely digested antigenic gluten proteins across an abnormally permeable intestinal epithelium. Many normal persons have increased IgA or IgG antigliadin.57 The frequency of elevated IgA or IgG DGP antibodies in healthy controls, however, is very low, possibly reflecting the antigenic potency of DGPs and their more central role in disease pathogenesis.14,54–56 Many persons with celiac disease have increased levels of serum antibodies against other food proteins, such as β-lactoglobulin, casein, and ovalbumin.58 It is unclear whether this reflects a general aberrant immune responsiveness to food antigens in patients with celiac disease or enhanced systemic exposure to these proteins because of increased small intestinal permeability. Gluten can be absorbed across normal epithelium, but it is unclear if this results in immune tolerance in persons who are not genetically predisposed to develop celiac disease.
The identification of more-specific autoantibody responses has altered our understanding of the pathogenesis of celiac disease. IgA antibodies to endomysium, a connective tissue structure surrounding smooth muscle, are virtually pathognomonic for celiac disease and are found only rarely in the absence of disease.59 It is now known that the target autoantigen contained within the endomysium is the enzyme tTG-2.12 Gliadin is a preferred substrate for this ubiquitous calcium-dependent intracellular enzyme, and it has been shown that tTG deamidates key neutral glutamine residues in gliadin and converts them into negatively charged glutamic acid residues, which are preferred in positions 4, 6, and 7 of the nonapeptide antigen-binding groove of the HLA-DQ2 heterodimer (see Fig. 104-4),13,14,60 thereby facilitating antigen presentation. Thus, tTG-mediated modification of gliadin to generate DGPs plays a pivotal role in eliciting a stronger proliferative response by gliadin-specific T-cell clones, or, stated differently, tTG makes gliadin tastier for the T cells.
With gliadin serving as the glutamine donor, tTG also can generate additional novel antigenic epitopes by cross-linking molecules of the extracellular matrix with gliadin or with tTG-gliadin complexes.61 As evidence of the fundamental role of tTG in celiac disease pathogenesis, one of the dominant epitopes responsible for the T-cell response contains a deamidated glutamine residue (Q65E) of α-gliadin.14 It also has been observed that tTG is necessary for the bioactivation of TGF-β that is required for epithelial differentiation. In a T84-crypt epithelial cell culture system, autoantibodies to tTG-blocked TGF-β–mediated enterocyte differentiation,62 a finding that suggests that release of tTG from cells during inflammation potentiates gliadin presentation by HLA-DQ2 and HLA-DQ8 and that local production of autoantibodies to tTG might contribute to the lack of epithelial differentiation observed in the active celiac lesion.
Given the marked infiltration of lymphocytes into the small intestinal mucosal epithelium and lamina propria in active disease, it is not surprising that cell-mediated immune responses also are important in the pathogenesis of celiac disease. Many findings support interplay between adaptive immunity, characterized by a specific and memory T-cell response to gluten peptides, and innate immunity, involving less-specific mechanisms. Many of the T cells in the small intestinal mucosa are activated in untreated celiac disease and release potent proinflammatory mediators such as IFN-γ, TNF-α, IL-2, IL-6, and TGF-β.48 Activated T lymphocytes, most of which are CD4+ cells, are abundant in the lamina propria of the small intestine.63 In contrast, IELs, which are present in large numbers in untreated celiac disease, are predominantly CD8+ T cells.64
There is an influx of primed memory T cells, marked by high CD45RO expression, in the mucosa of untreated celiac disease patients.65 In healthy persons, more than 90% of IELs express the α/β TCR, whereas expression of the γ/δ TCR by IELs in patients with untreated celiac disease is increased as much as six-fold (to 35%) and is considered a hallmark of the disease.66 These primitive lymphocytes recognize bacterial nonpeptide antigens and unprocessed stress-related proteins. They appear to act as mucosal guardians and might protect the intestinal mucosa from chronic exposure to dietary gluten in gluten-tolerant persons by secreting IL-4, which dampens Th1 in favor of Th2 reactivity.67 Their continuous presence in patients on a gluten-free diet might indicate inadvertent gluten ingestion. Patients with refractory celiac disease also have aberrant IELs with restricted γ/δ TCR gene rearrangements indicating oligoclonality. The pathogenetic role of these lymphocytes, compared with lamina propria lymphocytes, continues to evolve (see “Refractory Celiac Disease”).68
Studies suggest that IL-15 plays a key role in bridging the innate and adaptive immune responses in pathogenesis of celiac disease.15,16,69 This enterocyte- and macrophage-derived proinflammatory cytokine is increased massively in the mucosa of patients with active celiac disease and refractory celiac disease. Although the mechanisms that lead to its overproduction remain unknown, IL-15 regulates IEL homeostasis by promoting migration, preventing apoptosis, and enhancing the capacity of dendritic cells to function as antigen-presenting cells.69 In response to gliadin peptides, IL-15 triggers an adaptive CD4+ T-cell response in the lamina propria and also is capable of inducing direct epithelial cell injury by inducing IEL secretion of IFN-γ.16
CLINICAL FEATURES
Samuel Gee’s classic description, with its evocative account, was concerned largely with the gross manifestations of the disorder.5 This florid presentation, however, is now unusual in the Western world, constituting only the extreme tip of the celiac iceberg. Although some patients still present with severe illness, most have few, subtle, or no symptoms at diagnosis. Such cases may be identified by screening relatives of patients during research studies or from screening patients with associated disorders, such as type 1 diabetes mellitus, autoimmune thyroid disease, or Down syndrome. Incidental hematologic abnormalities (e.g., iron deficiency anemia) or biochemical abnormalities (e.g., elevated serum aminotransferase levels) also can lead to a diagnosis of celiac disease.
CHILDHOOD PRESENTATION
The classic presentation of celiac disease in infancy is not easily missed. The typical history is of steatorrhea with or without vomiting and occasional cramping abdominal pain that can occur anytime after weaning when cereals are introduced into the diet, but especially in the first and second years of life. Classically, the child fails to thrive, is apathetic and irritable, and has muscle wasting, hypotonia, and abdominal distention. Watery diarrhea or occasionally constipation may be reported. Diagnosis is more difficult when gastrointestinal features are less prominent, and the possibility of gluten sensitivity should be considered in all children who present with short stature or failure to thrive, even when there are no other symptoms to suggest an enteropathy. Once a gluten-free diet is commenced, catch-up growth is well documented.70 Nutritional deficiencies, particularly anemia, are another common mode of presentation, especially in older children. With earlier diagnosis, clinical rickets now is an uncommon complication but is seen occasionally, especially among Asian children with untreated celiac disease. Many pediatric patients enjoy a temporary, spontaneous remission of symptoms during adolescence, and it is unusual for celiac disease to manifest during the teens.
Considerable debate continues as to why celiac disease tends to be diagnosed later and with milder signs and symptoms than in the past. A number of studies suggest that breast-feeding can significantly delay the onset of symptoms,71,72 but not all studies support this conclusion.73 In one study of at-risk children, the introduction of gluten into the diet during the first three months of life or after seven months of age was associated with a significantly increased risk for celiac disease (hazard ratio [HR] 23.0; 95% confidence interval [CI]: 4.6-115.9; P = 0.001) compared with introduction at four to six months (HR 3.98; 95% CI: 1.18-13.46; P = 0.04).74
ADULTHOOD PRESENTATION
In the past, celiac disease was perceived to be a pediatric disorder, but the diagnosis now is being made increasingly in adults; currently, the overall mean age at presentation is approximately 45 years. Symptoms also have changed during the past 50 years. Diarrhea now is reported less often, and many patients now present with higher body mass indices and even with obesity. The unmasking of asymptomatic disease by surgery that induces rapid gastric emptying (e.g., gastric resection, pyloroplasty) or the finding of the typical lesion in asymptomatic relatives of celiac disease patients suggests that adults can have silent celiac disease for some time. A proportion of these adult patients have short stature or give a history consistent with unrecognized celiac disease in childhood. In many, however, there is nothing to suggest previous disease, and it is possible that celiac disease can develop for the first time in adult life. Celiac disease also is being diagnosed increasingly in later life, with approximately 25% of cases diagnosed in patients older than 60 years.75
GASTROINTESTINAL FEATURES
Several factors contribute to the diarrhea associated with celiac disease. The stool volume and osmotic load delivered to the colon are increased by the malabsorption of fat,76 carbohydrate, protein, electrolytes, and other nutrients. In addition, the delivery of excessive dietary fat into the large bowel results in the production by bacteria of hydroxy fatty acids, which are potent cathartics. Electrolytes actually are secreted into, rather than absorbed from, the lumen of the severely damaged upper small intestine in symptomatic patients. This secretion further increases luminal fluid in an intestine with an already compromised absorptive capacity. There also is evidence that secretin and cholecystokinin release in response to a meal are impaired in celiac disease, diminishing delivery of bile and pancreatic secretions into the gut lumen and possibly compromising intraluminal digestion.77 Alterations in the secretion of other intestinal peptides have been noted and can contribute to the observed diarrhea. Finally, if the disease extends to and involves the ileum, patients can experience the direct cathartic action of malabsorbed bile salts on the colon.76
Vague abdominal discomfort and especially abdominal bloating are extremely common and can lead to a mistaken diagnosis of irritable bowel syndrome (IBS). Because of the difficulty in distinguishing celiac disease with mild gastrointestinal manifestations from symptomatic IBS, serologic testing of IgA EMAs or IgA tTG should be considered in patients with symptoms suggesting diarrhea-predominant IBS. In a UK study, Sanders and colleagues78 evaluated 300 consecutive new patients who fulfilled Rome II criteria for IBS and 300 healthy age- and sex-matched controls for celiac disease using IgA AGA (antigliadin antibody), IgG AGA, and EMA; two controls (0.7%) (both EMA positive) and 14 IBS patients (4.6%) had celiac disease (P = 0.004; odds ratio [OR], 7.0; 95% confidence interval [CI]: 1.7-28.0). Severe abdominal pain can occur but is uncharacteristic in uncomplicated celiac disease; its occurrence can suggest the presence of complications such as intussusception, ulcerative jejunitis, or intestinal lymphoma. Abdominal distention with excessive amounts of malodorous flatus is a common complaint. Conversely, nausea and vomiting are uncommon in uncomplicated celiac disease. Recurrent severe aphthous stomatitis affects many celiac patients and may be their sole presenting complaint. It is important to exclude celiac disease in cases of recurrent aphthous stomatitis because a significant proportion of these patients respond well to dietary treatment.79
EXTRAINTESTINAL FEATURES
As patients with celiac disease get older, they tend to present with complaints not directly referable to the gastrointestinal tract. These extraintestinal symptoms and clinical findings often result from nutrient malabsorption and can involve virtually all organ systems (Table 104-1).80 Extraintestinal features, including anemia, osteopenia, neurologic symptoms, and menstrual abnormalities, often prove more distressing to the patient than do the gastrointestinal symptoms.
Table 104-1 Extraintestinal Manifestations of Celiac Disease
| MANIFESTATION | PROBABLE CAUSE(S) |
|---|---|
| Cutaneous | |
| Ecchymoses and petechiae | Vitamin K deficiency; rarely, thrombocytopenia |
| Edema | Hypoproteinemia |
| Dermatitis herpetiformis | Unknown |
| Follicular hyperkeratosis and dermatitis | Vitamin A malabsorption, vitamin B complex malabsorption |
| Endocrinologic | |
| Amenorrhea, infertility, impotence | Malnutrition, hypothalamic-pituitary dysfunction |
| Secondary hyperparathyroidism | Calcium and/or vitamin D malabsorption causing hypocalcemia |
| Hematologic | |
| Anemia | Iron, folate, vitamin B12, or pyridoxine deficiency |
| Hemorrhage | Vitamin K deficiency; rarely, thrombocytopenia due to folate deficiency |
| Thrombocytosis, Howell-Jolly bodies | Hyposplenism |
| Hepatic | |
| Elevated liver biochemical test levels | Unknown |
| Muscular | |
| Atrophy | Malnutrition due to malabsorption |
| Tetany | Calcium, vitamin D, and/or magnesium malabsorption |
| Weakness | Generalized muscle atrophy, hypokalemia |
| Neurologic | |
| Peripheral neuropathy | Deficiencies of vitamins such as vitamin B12 and thiamine |
| Ataxia | Cerebellar and posterior column damage |
| Demyelinating central nervous system lesions | Unknown |
| Seizures | Unknown |
| Skeletal | |
| Osteopenia | Malabsorption of calcium and vitamin D |
| Osteoarthropathy | Unknown |
| Pathologic fractures | Osteopenia |
Anemia
Anemia is a common manifestation of celiac disease in children and adults and usually is caused by impaired iron or folate absorption from the proximal intestine; in severe disease with ileal involvement, vitamin B12 absorption also is impaired. Patients with extensive disease can bleed into the skin or mucous membranes or can develop hematuria, epistaxis, or vaginal or gastrointestinal bleeding. Bleeding can aggravate pre-existing anemia and most often is caused by a coagulopathy resulting from impaired intestinal absorption of fat-soluble vitamin K. Evidence of hyposplenism of unknown cause, with thrombocytosis, deformed erythrocytes, and splenic atrophy, occurs in up to 50% of adults with celiac disease but only rarely is seen in children.81 In many patients, evidence of hyposplenism disappears with elimination of gluten from the diet.81
Osteopenia
Osteopenia is the most common complication of celiac disease, and its prevalence increases with age at diagnosis. More than 70% of patients with untreated celiac disease have osteopenia,82 and osteoporosis occurs in more than one quarter of all celiac disease patients.83 Osteopenia develops as a result of impaired calcium absorption (secondary to defective calcium transport by the diseased small intestine), vitamin D deficiency (caused by impaired absorption of this fat-soluble vitamin), and binding of intraluminal calcium and magnesium to unabsorbed dietary fatty acids (forming insoluble soaps, which are then excreted in the feces). Chronic intestinal inflammation also can contribute to bone loss through release of inflammatory mediators.
Osteopenia is less common in patients with silent celiac disease, in whom prevalence rates between 30% and 40% have been reported.84 Whereas bone disease generally is more severe among patients with symptomatic disease, severe osteopenia has been reported in up to one third of symptom-free adults whose celiac disease was diagnosed during childhood and who resumed a normal diet during adolescence.85
A key unanswered question is the functional consequence of osteopenia. An increased risk of fractures was observed in patients with overt celiac disease in one study84 but not in another.86 The fracture risk among patients with silent celiac also remains unclear.
Neurologic Symptoms
Neurologic symptoms caused by lesions of the central or peripheral nervous system occasionally occur in patients with celiac disease and are poorly understood. Celiac disease often is found in patients presenting with nonhereditary ataxia, and progressive gait and limb ataxia may be the sole manifestations of disease in some patients. These abnormalities, referred to as gluten ataxia, are believed to result from immunologic damage to the cerebellum, posterior columns of the spinal cord, and peripheral nerves.87 Muscle weakness and paresthesias with sensory loss also are encountered occasionally, and pathologic evidence of peripheral neuropathy and patchy demyelinization of the spinal cord, cerebellar atrophy, and capillary proliferation suggestive of Wernicke’s encephalopathy have been described rarely.
Although potential causative roles for specific vitamin deficiencies (including vitamin B12, thiamine, riboflavin, and pyridoxine) have not been established, neurologic symptoms have been reported to improve in some patients who are given multivitamins, including vitamins A, B, and E, and calcium. Night blindness is a clear indication for vitamin A therapy. Peripheral neuropathy and ataxia, however, often appear unrelated to specific vitamin deficiency states and usually do not respond to gluten withdrawal.88
The associations of celiac disease and epilepsy, frequently complex partial seizures, and bilateral parieto-occipital cerebral calcification are well recognized.89 In one series, epilepsy was reported in approximately 5% of children and young adults with celiac disease.90 The cause of the epilepsy remains unclear, and the prognosis might depend on how early in the course of the disease a gluten-free diet is started.
Although most patients with celiac disease do not appear psychologically abnormal, many affected subjects report a striking improvement in mood after commencing a gluten-free diet.91
Gynecologic and Fertility Problems
Gynecologic and obstetric problems are common in women with untreated celiac disease.92 Amenorrhea occurs in one third of women of childbearing age and menarche is often delayed, typically by one year, in untreated subjects. Women with untreated celiac disease can present with infertility, and it is common for infertile women with celiac disease to become pregnant shortly after commencing a gluten-free diet.93
A high prevalence of silent celiac disease has been reported in women with recurrent spontaneous abortions, intrauterine fetal growth retardation, and unfavorable outcomes of pregnancy, underlining the need to test for celiac disease in these situations.94
Infertility secondary to impotence or an abnormally low sperm count can occur in men with untreated celiac disease.95 Although malnutrition, including folate deficiency related to malabsorption, can contribute to male infertility, abnormalities in hypothalamic-pituitary regulation of gonadal function and gonadal androgen resistance that disappears on gluten withdrawal also have been incriminated.95
Physical Examination
Examination of the mouth may show aphthous stomatitis, angular cheilosis, and glossitis with decreased papillation of the tongue. Dental enamel defects are common.96 The abdomen may be protuberant and tympanitic, with a characteristic doughy consistency, owing to distention of intestinal loops with fluid and gas. Hepatomegaly and abdominal tenderness are uncommon, but ascites may be detected in patients with significant hypoproteinemia. Peripheral lymphadenopathy is unusual in the absence of complicating lymphoma.








