Fig. 12.1
General scheme of ammonia metabolism in the liver . Solid arrows, pathways of ammonia utilization; broken arrows, pathways of ammonia formation (Reproduced with permission from Ampola [1])
The urea cycle is subject to both short- and long-term regulation. Short–term regulation is accomplished by intracellular substrate levels. In long–term regulation, total hepatic content of the five urea cycle enzymes is proportional to protein intake and rate of urea excretion. All five urea cycle enzymes may be induced by glucagon and glucocorticoids . The rate-limiting enzyme in vivo is probably carbamylphosphate synthetase I.
Ammonia is toxic to the central nervous system , and in disorders characterized by hyperammonemia, there is increased movement of ammonia across the blood–brain barrier. The cells most susceptible to ammonia toxicity are the astrocytic or glial cells. Astrocytes are the site of ammonia detoxification in the central nervous system with glutamine production from glutamate, a major excitatory neurotransmitter in the brain. In fact, cerebrospinal fluid glutamine levels parallel the severity of neurologic dysfunction. As the level of glutamate declines, it is replenished by condensation of ammonia with α-ketoglutarate. Depletion of α-ketoglutarate results in impaired mitochondrial glucose oxidation. Ammonia may also adversely affect neurotransmitter function and directly interfere with cellular energy production. As the degree of hyperammonemia increases, neurologic function progressively deteriorates with onset of deepening coma and finally development of cerebral edema, which is often a terminal event.
Inherited defects in the urea cycle enzymes may result in hyperammonemia and neurologic dysfunction, often to a severe degree. Inherited defects in all five enzymes of the cycle, as well as N-acetylglutamate synthetase (which produces an essential cofactor for carbamyl phosphate synthetase), have been described. There may be a spectrum of severity, depending on the specific defect in the enzyme and whether there is partial residual function. In severe cases, seizures, coma, and death may occur in the neonatal period, if untreated.
3 Hepatic Encephalopathy
Hepatic encephalopathy may be classified as two general types. In patients with cirrhosis , portal hypertension, and large portosystemic shunts, ammonia from intestinal sources is not cleared effectively on first-pass through the liver . Also, the relief of portal hypertension by surgical creation of a portosystemic shunt or placement of a transjugular intrahepatic portosystemic shunt (TIPS) may precipitate encephalopathy. This ineffective clearance, combined with generally poor hepatic function, may result in chronic encephalopathy, punctuated by acute exacerbations often precipitated by gastrointestinal bleeding from esophageal varices (resulting in an increased intestinal ammonia load), infection, excessive protein intake, or overzealous diuresis to treat ascites . Chronic pathologic changes in astrocytes in the central nervous system may be present. This type of encephalopathy is referred to as portosystemic encephalopathy . The other category of encephalopathy is observed in the patient with acute, severe liver disease (usually fulminant hepatitis ) who develops progressive encephalopathy due primarily to severe liver dysfunction with inadequate ammonia detoxification . Acute liver failure with encephalopathy often rapidly progresses to cerebral edema and death. The cerebral edema is thought to be a direct result of ammonia toxicity. In fact, the edema may actually be a result of the osmotic effect of ammonia-induced elevation of brain glutamine levels.
It should be stressed that in encephalopathy associated with liver disease, other factors may contribute to the central nervous system dysfunction in addition to hyperammonemia. In fact, the degree of hyperammonemia does not always correlate with the degree of encephalopathy, suggesting that other mechanisms may be involved. In acute liver failure, other neurotoxic substances, such as mercaptans and short–chain fatty acids , may play a role. There is evidence that hepatic encephalopathy may result in part from the action of endogenous substances that bind to the GABA–benzodiazepine receptor complex in the brain. This is supported clinically by the observation that portosystemic encephalopathy and encephalopathy associated with acute liver failure may improve transiently with treatment with benzodiazepine receptor antagonists. However, there are limited data to support their clinical use. There may be increased entry of ammonia into the central nervous system due to increased permeability of the blood–brain barrier and conversion of positively charged ammonium ion to ammonia, which more readily crosses the blood–brain barrier, secondary to changes in blood pH. Accumulation of glutamine in the brain results in enhanced central nervous system uptake of tryptophan , which is converted into both neuroactive and neurotoxic metabolites, including serotonin and tryptamine.
Clinically, the presentation of the first stage of hepatic encephalopathy may be subtle. There may be personality changes and inversion of sleep patterns. The patient may exhibit a short attention span and easy forgetfulness. With portosystemic encephalopathy , it is now recognized that very mild impairment may affect social skills and emotional behavior, and evaluation of driving skills may be a practical index to diagnose what is termed minimal hepatic encephalopathy (MHE). An early physical sign of hepatic encephalopathy is asterixis. This sign represents an inability to maintain posture and is usually elicited by asking the patient to dorsiflex the hand with the arm extended. After several seconds a positive sign is characterized by the hand falling forward followed by a brisk recovery, thereby resulting in the typical “flapping” tremor. This sign is dependent upon the patient being able to understand and follow directions, and is highly variable over short periods of time, even in the same individual. Asterixis is not specific for hepatic encephalopathy and may be elicited in other disorders, including uremia. In stage 2, the individual becomes lethargic and disoriented with other abnormal reflexes in addition to asterixis. In stage 3, the patient is somnolent, but arousable. However, when awake the patient is unable to communicate or perform mental tasks such as calculation. Stage 4 encephalopathy is characterized by total inability to arouse the patient accompanied by a positive Babinski sign and decerebrate posturing, and usually immediately precedes the development of cerebral edema and death. Abnormal changes on electroencephalogram (EEG) are detectable as early as stage 1 and generally worsen in parallel with clinical progression to stage 4.
Treatment of hyperammonemia should be directed toward reducing the production of ammonia and enhancing its clearance. Moderate protein restriction had been recommended in the past. However, since restriction results in enhanced catabolism of endogenous protein, and a significant positive effect on outcomes has yet to be proven, such restriction is not currently recommended. Branched–chain amino acid supplementation may have a modest beneficial effect, but is prohibitively expensive. Attention should be directed to sources of gastrointestinal bleeding, such as esophageal varices , which may result in a large intestinal ammonia load. Lactulose is a non-absorbable carbohydrate administered enterally and is fermented by colonic bacteria to form organic acids, which lower the luminal pH. Since only ammonia in the form of NH3 is readily absorbed, an increase in the NH4 +/NH3 ratio results in trapping of ammonia in the gut and elimination in the feces. Also, the osmotic property of lactulose results in a significant laxative effect and may effectively clear ammoniagenic substances from the gastrointestinal tract. The non-absorbable antibiotic, rifaximin , is used to suppress intestinal ammonia-forming bacteria. In urgent situations, high ammonia levels may also be cleared by hemodialysis or exchange transfusion. Use of external liver support systems has generally not significantly impacted outcomes. The molecular absorbent recirculating system (MARS) has shown some benefit in more severe cases. In cases of encephalopathy associated with liver failure, liver transplantation may result in prompt resolution of severe encephalopathy, providing that cerebral edema has not developed.
4 Hepatic Detoxification
The liver is responsible for the biotransformation and clearance of a variety of endogenous and exogenous toxic compounds. Exogenous organic substances entering the body that have no nutritive value for energy production, structural function, or enzyme cofactor function are called xenobiotics. The liver is capable of numerous reactions which generally serve to make the substance to be eliminated more polar and water soluble to subsequently be excreted in bile or urine (Phase I reactions: oxidoreductases and hydrolases) and to make the substance less toxic (Phase II reactions: transferases). As will also be demonstrated, these reactions do not always result in detoxification , but may actually form toxic intermediary metabolites. Apical canalicular excretion of these biotransformed molecules into bile for elimination or basolateral membrane transport into the circulation to be cleared by the kidney occurs via specific ATP–binding cassette (ABC) transporters and is termed Phase III transport.
4.1 Phase I Reactions
The enzyme systems responsible for oxidation and reduction reactions are components of Phase I biotransformation. Dehydrogenases catalyze hydrogen transfer from a substrate to a hydrogen receptor, frequently a pyridine nucleotide. Examples include alcohol dehydrogenases, cytosolic enzymes, which catalyze NAD-dependent oxidation of alcohols to aldehydes or ketones by the following reaction:
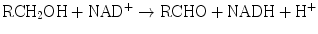
Aldehyde dehydrogenases are present in mitochondrial, microsomal, and cytosolic hepatocyte fractions. The production of aldehydes from alcohols in the above reaction is reversible. However, conversion of the aldehydes to ketones by aldehyde dehydrogenases is essentially irreversible:
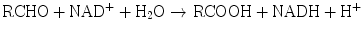
Another dehydrogenase is dihydrodiol dehydrogenase, which catalyzes the NADP+-dependent oxidation of metabolites of polycyclic aromatic hydrocarbon compounds.
Hepatic reductases include aldehyde and ketone reductases, quinone reductases, nitro and nitroso reductases, azoreductases, N–oxide reductases, and sulfoxide reductases. Substrates for these enzymes are drugs and xenobiotics, including warfarin , aflatoxin, adriamycin, chloramphenicol, and dimethylsulfoxide. Oxidases catalyze transfer of electrons from substrate to oxygen, generating either hydrogen peroxide or superoxide anions, and include aldehyde oxidases and monoamine oxidases.
The hepatic mono–oxygenases are microsomal enzymes that catalyze reactions in which one atom of oxygen is incorporated into product and the other is reduced to water:
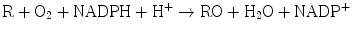
The cytochrome P450–dependent mono–oxygenases have been the most intensively studied. Although these enzymes are found in many tissues throughout the body, they are most abundant in the liver . Within the hepatocyte this system is located primarily in the smooth endoplasmic reticulum. It is also found in the nuclear membrane, where this fraction is probably more important in carcinogenesis. The system derives its name from “P” which stands for “pigment” and “450” which indicates the absorption wavelength in nm at which a maximum occurs with the binding of carbon monoxide under anaerobic conditions. This mono-oxygenase system consists of a flavoprotein and a family of hemoproteins. The scheme for the function of this complex is shown in Fig. 12.2. In the first step, the oxidized (Fe3+) form of the enzyme binds the reduced substrate. In the rate–limiting second step, the complexed cytochrome is reduced to the Fe2+ form by a flavoprotein, NADPH-cytochrome P450 reductase, with NADPH as the ultimate electron donor. In step three, the reduced cytochrome-drug complex combines with molecular oxygen to yield a ternary intermediate in which the iron is again oxidized to the Fe3+ state. In the fourth step, the ternary complex decomposes to yield the free oxidized cytochrome, the oxidized form of the substrate, and water. The entire cycle involves a two–electron transfer and consumption of one mole of NADPH and one mole of molecular oxygen per mole of drug reduced. The components of this system are integral membrane proteins. Reconstitution of the proteins in vitro requires the presence of phospholipid for a catalytically active complex. These mono-oxygenases are subject to genetic variation and can be induced or inhibited by xenobiotics, which may affect the metabolism of certain drugs.
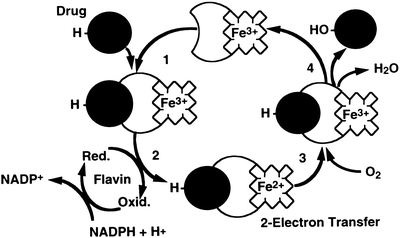
Fig. 12.2
The cytochrome P450-dependent mono-oxygenase redox cycle. In the first step, the oxidized (Fe3+) form of the enzyme binds the reduced drug substrate. In the rate-limiting second step, the complexed cytochrome is reduced to the Fe2+ form by a flavoprotein, NADPH-cytochrome P450 reductase, with NADPH as the ultimate electron donor. In step three, the reduced cytochrome-drug complex combines with molecular oxygen to yield a ternary intermediate in which the iron is again oxidized to the Fe3+ state. In the fourth step, the ternary complex decomposes to yield the free oxidized cytochrome, the oxidized form of the drug, and water
Hydrolytic reactions involve substrates that contain ester linkages. Hydrolases cleave ester linkages with addition of a molecule of water:
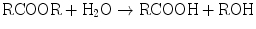
The products of this hydrolysis, an acid and an alcohol, are more water-soluble than the parent ester. In addition to increasing water solubility, this reaction often results in detoxification of the xenobiotic. The reaction products may also be substrates for Phase II reactions. Xenobiotics subject to hydrolysis include aspirin, meperidine, malathion, and nerve gas.
4.2 Phase II Reactions
The general scheme for conjugation reactions is as follows:
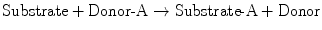
One of the most common conjugation reactions is glucuronidation . Glucuronic acid is transferred from UDP–glucuronic acid (UDPGA) to the hydroxyl or carboxyl group on the acceptor substrate molecule to form a β-glucuronide. Glucuronidation generally not only enhances water solubility, but also detoxifies the compound. Substrates for this reaction include acetaminophen, morphine, chloramphenicol, furosemide, sulfisoxazole, and valproic acid. As discussed subsequently, bilirubin, an endogenous waste product of heme catabolism, is a major substrate for conjugation with glucuronide. The glucuronidation capacity of the liver depends on the rate of synthesis of UDPGA from UDP-glucose , which requires adequate carbohydrate reserves. Another Phase II conjugation reaction is sulfation. Conjugation with sulfate generally results in compounds that are less toxic and more water-soluble. Substrates for this reaction include alcohols, bile acids, and phenolic compounds.
Glutathione (GSH) is a tripeptide (γ-glutamyl-cysteinyl-glycine ) which participates in substitution reactions that are catalyzed by glutathione S–transferases, whereby the GSH displaces a group on a xenobiotic, which generally has an electrophilic center. Electrophilic compounds are particularly toxic to cells because of their propensity to react spontaneously with -SH groups on proteins or amine groups on nucleic acids, leading to cell necrosis or carcinogenesis. Substrates for this reaction include nitroglycerin, the diuretic ethacrynic acid, certain insecticides, and sulfobromophthalein (BSP). The GSH conjugates are generally excreted into bile. However, they may be reabsorbed in the intestine and subsequently taken up by the kidney, where the enzyme γ-glutamyl–transpeptidase (GGTP) transfers the γ-glutamyl group of the conjugate to an amino acid, leaving the cysteine-glycine conjugate. A dipeptidase then removes the glycine, producing the cysteine conjugate. This conjugate may be excreted in the urine or acetylated to mercapturic acid in the liver or kidney. The mercapturic acids may be excreted in urine, and to a lesser extent, in bile. Under certain conditions, such as acetaminophen overdosage to be discussed subsequently, the GSH stores may be depleted and the potential for toxicity greatly increases.
Methylation is another Phase II reaction, which serves mainly to detoxify and actually may render some substrates less water-soluble. In this reaction the methyl group of S-adenosylmethionine is transferred to an acceptor xenobiotic that contains a polyphenol, amine, or sulfhydryl group. Substrates for this reaction include norepinephrine , epinephrine , dopamine, and tyramine and other catachols after hydroxylation by the cytochrome P450 system.
5 Bioactivation and Hepatocellular Injury
Although the biotransformation reactions just described generally function to detoxify and enhance the elimination of xenobiotics, there are instances in which the substrates are actually rendered more toxic or carcinogenic. There are five classes of such biotransformation. The xenobiotic may be transformed into a stable, but toxic metabolite. Examples include acetonitrile and methoxyflurane. A second class involves transformation into a reactive, electrophilic metabolite. Acetaminophen and furosemide are in this class. Third, the substrate may be converted to a free radical, as in the case of carbon tetrachloride. Fourth, the biotransformation reaction may lead to the formation of reduced oxygen metabolites, as occurs with paraquat and quinones. Finally, the biotransformation reaction itself may generate a metabolic derangement within the cell, as in the case of galactosamine–induced liver injury, where there is a depression of uracil nucleotide dependent biosynthesis of nucleic acids, glycolipids, glycoproteins , and glycogen, which results in cell organelle injury.
Review of the biochemical events occurring in acetaminophen toxicity serves as an excellent example of these metabolic pathways. These events are summarized in Fig. 12.3. When taken in appropriate doses, most of the acetaminophen is conjugated with sulfate and glucuronic acid and eliminated. About 5-15 % is converted to a reactive, electrophilic intermediate, N–acetyl–p–benzoquinone imine (NAPQI), which reacts with sulfhydryl groups in proteins. The NAPQI is normally immediately conjugated with glutathione before it can do any damage to the cell and is converted into a mercapturic acid for mainly renal elimination. However, in the case of an overdose the conjugation pathway is saturated and available glutathione is quickly depleted, allowing accumulation of significant amounts of NAPQI. The NAPQI causes cell injury and death resulting in the hepatic necrosis seen with acetaminophen overdose, which can result in liver failure and death. Obviously, one therapeutic strategy would be repletion of the liver’s glutathione stores. Although this cannot be done directly, another successful approach is to administer N–acetylcysteine (Mucomist R ), which restores glutathione levels by enhancing synthesis. If administered early enough in the course of intoxication, this intervention can effectively prevent significant liver injury.
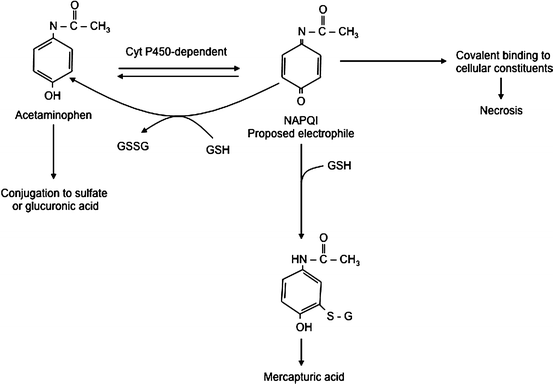
Fig. 12.3
Hepatic metabolic pathways in acetaminophen overdose. GSH glutathione, NAPQI N-acetyl-p-benzoquinone imine (Reproduced with permission from Vessey [20])
6 Hepatic Bilirubin Metabolism
Bilirubin is a pigment derived from the catabolism of heme. It is a toxic waste product, which undergoes hepatic biotransformation and excretion in bile. Bilirubin has an open–chain tetrapyrrole structure with eight side chains (Fig. 12.4). The IX-α isomer is the most abundant naturally occurring isomer of bilirubin. From the chemical structure of bilirubin, one would predict a water-soluble compound at physiological pH. However, in reality, unconjugated bilirubin has only very limited water solubility. This phenomenon may be explained by the presence of extensive internal hydrogen bonding within the molecule as shown in Fig. 12.5. This results in the involvement of all of the polar groups of the molecule in the hydrogen bonding, so that none are available for hydration.
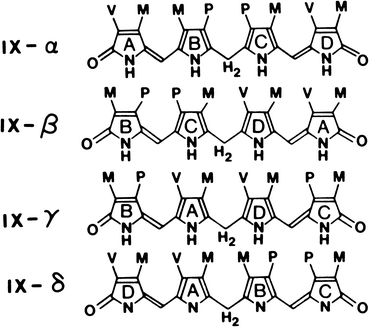
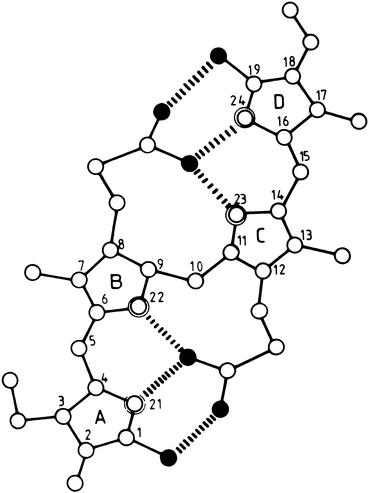
Fig. 12.4
The four isomeric forms of bilirubin. The IX-α isomer is the most abundant naturally occurring form. (M, CH3; V, CH2 = CH2; P, CH2-CH2-COOH.) (Reproduced with permission from Odell [14])
Fig. 12.5
Molecular structure of internally hydrogen-bonded bilirubin IX-α. Open, double-contoured, and filled circles represent carbon, nitrogen, and oxygen atoms, respectively. Hydrogen atoms are not shown. The six intramolecular hydrogen bonds are indicated by broken lines (Reproduced with permission from Blanckaert and Fevery [2])
Normally, serum bilirubin levels are very low, and unconjugated bilirubin predominates. In normal individuals, men have slightly higher serum bilirubin levels (0.6 mg/dL) than females (0.4 mg/dL), and any level above 1.0 mg/dL is considered abnormal. Jaundice with the appearance of the pigment in sclerae, skin, and mucus membranes generally is noted when the bilirubin level exceeds 3 mg/dL in adults and 6 mg/dL in infants.
The major site of bilirubin toxicity is the central nervous system , particularly in the newborn infant. Bilirubin inhibits central nervous system RNA and protein synthesis, uncouples oxidative phosphorylation, and inhibits ATPase activity in brain mitochondria. Clinically, bilirubin toxicity can produce severe central nervous system dysfunction resulting in permanent injury or death. Accumulation of bilirubin in the brain, primarily in the basal ganglia, hippocampus, and cochlear and cerebellar nuclei, with associated toxicity is termed kernicterus .
Bilirubin is the major degradation product of heme in mammals. Degradation of hemoglobin from senescent red blood cells by reticuloendothelial cells in the spleen accounts for 80 % of bilirubin production. The other 20 % is derived from ineffective erythropoiesis in the bone marrow, as well as the turnover of heme-containing proteins and myoglobin. The series of reactions responsible for the conversion of heme into bilirubin is shown in Fig. 12.6. The first step is the conversion of heme to biliverdin mediated by the enzyme heme oxygenase, which exists as three isoforms. The activity of this microsomal enzyme is highest in the spleen and Kupffer cells in the liver and requires NADPH and oxygen. This is the only reaction in the body that produces carbon monoxide, which is excreted in expired air and may be quantitated as an index of bilirubin production. Carbon monoxide is also an important signaling molecule and functions as a potent vasodilator. Interestingly, biliverdin is a non-toxic, water-soluble compound and is the end-product of heme catabolism in birds and other non-mammalian vertebrates. However, in mammals biliverdin is converted to bilirubin, which is toxic and requires energy-dependent biotransformation for excretion. Conversion of biliverdin to bilirubin also occurs in reticuloendothelial cells by biliverdin reductase, a cytosolic enzyme that requires NADPH, and utilizes NADH at lower pH. Both bilirubin and biliverdin may have beneficial effects as anti–oxidants under certain conditions and may affect progression of atherosclerosis.
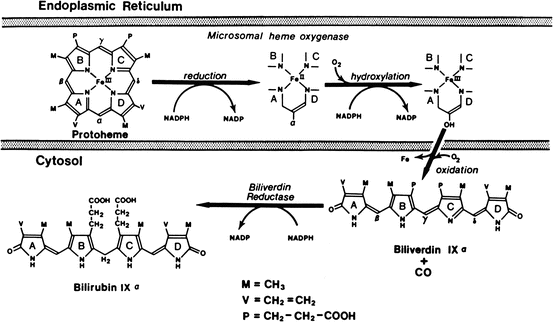
Fig. 12.6
Pathways of biliverdin production from heme by chain-opening and bilirubin production by reduction of biliverdin (Reproduced with permission from Odell [15])
After its formation in reticuloendothelial cells, bilirubin is released into the bloodstream, where it is transported bound to albumin via one primary and two secondary binding sites. This binding prevents the diffusion of free bilirubin into tissues, particularly the central nervous system , thereby preventing toxicity. However, other ligands such as drugs (salicylates, sulfonamides, furosemide, etc.) and fatty acids can displace bilirubin from albumin binding sites and precipitate toxicity.
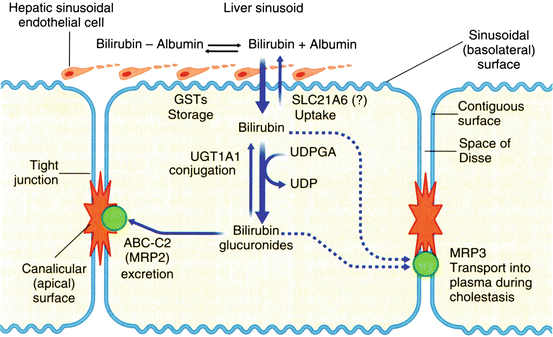
At the hepatocyte plasma membrane, bilirubin enters the hepatocyte via a specific uptake mechanism, as shown in Fig. 12.7. Once inside the cell, bilirubin binds to a group of cytosolic carrier proteins, glutathione–S–transferases (GSTs), also termed ligandin or Y protein, keeping the bilirubin in solution and within the cell. The bilirubin is then conjugated with glucuronide by the microsomal enzyme, bilirubin UDP–glucuronosyltransferase (UGT1A1). The resulting bilirubin monoglucuronide and diglucuronide are excreted at the canalicular membrane as a component of bile by the ATP–dependent transporter MRP2 (ABCC2). The transcription factors, constitutive androstane receptor (CAR) and pregnane X receptor (PXR), regulate the various proteins involved in bilirubin uptake, glucuronidation and secretion. Conjugation renders bilirubin non–toxic, water–soluble, and non–absorbable from the gastrointestinal tract. In the distal small intestinal and colonic lumen, the conjugated bilirubin is converted to urobilinogens by bacteria and eliminated in stool. However, a small fraction of these tetrapyrroles may be reabsorbed and re-excreted in bile, and the kidney excretes a small fraction.
 < div class='tao-gold-member'>
< div class='tao-gold-member'>




Only gold members can continue reading. Log In or Register to continue
Related posts:






Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
