Although androgen-deprivation therapy is the standard therapy for advanced and metastatic prostate cancer, this treatment is only palliative. Prostate cancer recurs then grows despite low circulating testicular androgens, using several mechanisms that remain dependent on androgen-receptor signaling in most cases. This article reviews the diversity of mechanisms used for growth by castration-recurrent prostate cancer.
- •
Castration-recurrent prostate cancer is androgen-stimulated.
- •
The clinical terms hormone-refractory and androgen-independent are obsolete and should be abandoned.
- •
Castration-recurrent prostate cancer is androgen stimulated, and remains dependent on androgen-receptor signaling in most cases.
- •
Several mechanisms are used by castration-recurrent prostate cancer cells to grow despite castrate serum androgens levels. These mechanisms include the hypersensitive, the intracrine androgen synthesis, the promiscuous, the outlaw, the bypass, and the stem-cell pathways.
- •
Many promising therapeutic agents are becoming available for treatment of castration-recurrent prostate cancer because of better understanding of the molecular processes that fuel the growth of castration-recurrent prostate cancer.
Introduction
Prostate cancer (CaP) is the second leading cause of death from cancer in American men. Almost all cases of CaP mortality occur as a result of metastatic disease. More than 7 decades ago, Huggins and Hodges demonstrated the benefits of androgen-deprivation therapy (ADT) for metastatic CaP. On initiation of ADT, most men experience significant clinical, biochemical, and radiologic remission. However, this response is only temporary and almost all patients experience disease recurrence, hence the term castration-recurrent (castration-resistant) CaP (CRCaP), which is fatal in 80% of patients. Until recently, CRCaP was inaccurately labeled as hormone-refractory or androgen-independent. However; recent advances have shown that CRCaP remains dependent on androgen receptor (AR) signaling in most cases. This article reviews the diversity of mechanisms used for growth by CRCaP ( Table 1 ).
| Pathway | Ligand Availability | AR Dependence | Components |
|---|---|---|---|
| Hypersensitive | Testicular androgens | Yes |
|
| Intracrine testicular androgen synthesis | Testicular androgens | Yes |
|
| Promiscuous | Testicular and adrenal androgens, corticosteroids, estradiol, progesterone, antiandrogens | Yes | Widened AR specificity via AR mutations or altered coregulator profiles |
| Outlaw | No | Yes |
|
| Bypass | No | No |
|
| Stem cell | No | No | Prostate cancer stem cells |
The androgen receptor
The biological effects of androgens are mediated through AR, a member of the nuclear steroid hormone receptor superfamily. The AR gene is located on the X chromosome (Xq11–12), spans approximately 180 kb of DNA, and has 8 exons (exons A–H). The AR protein is composed of 919 amino acids and is divided into 4 domains: an amino-terminal transactivation domain (NTD), a DNA-binding domain (DBD), a hinge region, and a ligand-binding domain (LBD) ( Fig. 1 ).
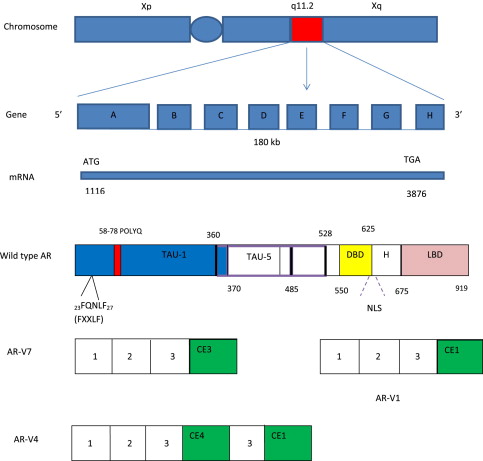
The AR NTD, encoded by exon A, constitutes about half of the AR protein and is responsible for the majority of the AR transcriptional activities and AR coactivator interaction. The AR NTD is composed of 2 transcription activation units (TAU): TAU-1 (amino acids 1–485), required for full ligand-stimulated, wild-type AR activity, and TAU-5 (amino acids 360–528), which confers constitutive ligand-independent AR activity. Unique to AR is the variable-length polyglutamine repeat (polyQ) in the NTD. Shorter polyQ appears to increase AR transcriptional activity, and has been linked to increased CaP incidence and aggressiveness in some but not other studies. Long polyQ (>40) causes spinal bulbar muscular atrophy (Kennedy disease).
The AR DBD is composed of 2 zinc-finger motifs, which are encoded by exons B and C, respectively. The first zinc finger mediates DNA recognition through interaction with specific base pairs in response elements, which facilitates binding of AR to the major groove of DNA. The second zinc finger stabilizes DNA binding and mediates AR dimerization. The hinge region, encoded by part of exon D, contains a bipartite nuclear localization signal as well as important sites for phosphorylation, acetylation, and degradation. The LBD, encoded by part of exon D and exons E to H, mediates ligand binding. The AR LBD has 12 conserved α-helices that form a ligand-binding pocket. Agonist binding induces a conformational change in the LBD and the folding of helix 12 across the ligand-binding pocket, which forms the activation function 2 (AF2) surface. The AF2 surface mediates interaction with the FQNLF peptide of the AR NTD and can bind coregulators containing similar sequences. In the absence of ligand binding, AR is present diffusely throughout the cytoplasm and held in an inactive state in association with chaperones, such as heat-shock proteins (HSPs). The minimal chaperone complex required for efficient folding and stabilization of AR consists of Hsp70 (hsc70), Hsp40 (Ydj1), Hop (p60), Hsp90, and p23. Ligand binding releases AR from HSPs, and is followed by nuclear translocation, homodimer formation, binding to AR response element DNA (target genes), and recruitment of AR coregulators (coactivators and/or corepressors), which regulate gene transcription. The best characterized AR target gene is Kallikrein-related peptidase 3 gene (KLK3), which encodes the prostate-specific antigen (PSA) protein.
The androgen receptor
The biological effects of androgens are mediated through AR, a member of the nuclear steroid hormone receptor superfamily. The AR gene is located on the X chromosome (Xq11–12), spans approximately 180 kb of DNA, and has 8 exons (exons A–H). The AR protein is composed of 919 amino acids and is divided into 4 domains: an amino-terminal transactivation domain (NTD), a DNA-binding domain (DBD), a hinge region, and a ligand-binding domain (LBD) ( Fig. 1 ).
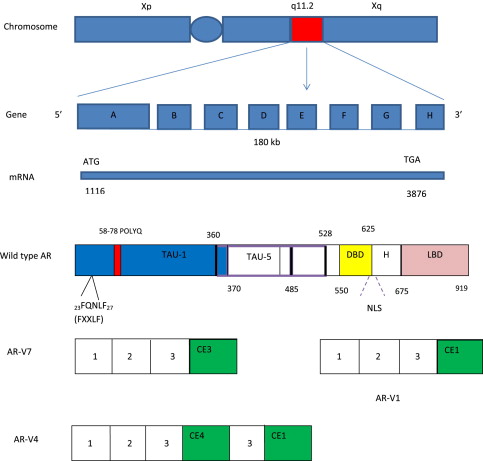
The AR NTD, encoded by exon A, constitutes about half of the AR protein and is responsible for the majority of the AR transcriptional activities and AR coactivator interaction. The AR NTD is composed of 2 transcription activation units (TAU): TAU-1 (amino acids 1–485), required for full ligand-stimulated, wild-type AR activity, and TAU-5 (amino acids 360–528), which confers constitutive ligand-independent AR activity. Unique to AR is the variable-length polyglutamine repeat (polyQ) in the NTD. Shorter polyQ appears to increase AR transcriptional activity, and has been linked to increased CaP incidence and aggressiveness in some but not other studies. Long polyQ (>40) causes spinal bulbar muscular atrophy (Kennedy disease).
The AR DBD is composed of 2 zinc-finger motifs, which are encoded by exons B and C, respectively. The first zinc finger mediates DNA recognition through interaction with specific base pairs in response elements, which facilitates binding of AR to the major groove of DNA. The second zinc finger stabilizes DNA binding and mediates AR dimerization. The hinge region, encoded by part of exon D, contains a bipartite nuclear localization signal as well as important sites for phosphorylation, acetylation, and degradation. The LBD, encoded by part of exon D and exons E to H, mediates ligand binding. The AR LBD has 12 conserved α-helices that form a ligand-binding pocket. Agonist binding induces a conformational change in the LBD and the folding of helix 12 across the ligand-binding pocket, which forms the activation function 2 (AF2) surface. The AF2 surface mediates interaction with the FQNLF peptide of the AR NTD and can bind coregulators containing similar sequences. In the absence of ligand binding, AR is present diffusely throughout the cytoplasm and held in an inactive state in association with chaperones, such as heat-shock proteins (HSPs). The minimal chaperone complex required for efficient folding and stabilization of AR consists of Hsp70 (hsc70), Hsp40 (Ydj1), Hop (p60), Hsp90, and p23. Ligand binding releases AR from HSPs, and is followed by nuclear translocation, homodimer formation, binding to AR response element DNA (target genes), and recruitment of AR coregulators (coactivators and/or corepressors), which regulate gene transcription. The best characterized AR target gene is Kallikrein-related peptidase 3 gene (KLK3), which encodes the prostate-specific antigen (PSA) protein.
The hypersensitive pathway
CRCaP can increase its sensitivity to available low androgen levels by increasing AR synthesis and/or increasing AR sensitivity.
AR Gene Amplification and AR Overexpression
AR gene amplification was first reported in 1995. Early studies reported AR gene amplification in 28% to 30% of CRCaP patients. AR gene amplification seemed important because no AR gene amplification was found in primary CaP in eugonadal patients (androgen-stimulated CaP [ASCaP]). AR gene amplification was detected mainly in CRCaP that had initially responded well to ADT and whose response duration was more than 12 months. CaP that recurred earlier or failed to respond to ADT had no AR gene amplification. Median survival after CaP recurrence was twice as long for patients with AR gene amplification as for those with no amplification ( P = .03). A subsequent report from the same group that examined prostatic tissues from a larger cohort of CRCaP patients failed to detect an association between AR gene amplification and survival.
Our group detected AR gene amplification in 33% of CRCaP specimens obtained from the primary site, which was associated with increased AR protein expression. However, no association was found between AR gene amplification and duration of survival after ADT. More recently, AR gene amplification was detected in 50% to 85% of circulating tumor cells in patients with CRCaP. These differences in reported rates of AR overexpression could be attributed to study cohort differences, tumor heterogeneity, and technical improvements.
Holzbeierlein and colleagues performed a genome-wide analysis and identified gene expression changes that occur during ADT and in CRCaP. Many AR-regulated genes that were suppressed when patients were started on ADT were reexpressed in CRCaP, specifically AR and steroid-synthesis enzymes, which suggests that CRCaP tumors have increased sensitivity to, and endogenously synthesize androgens (intracrine androgen synthesis; see later discussion). Chen and colleagues selected 7 CaP xenografts for castration resistance by passing tumors in castrated mice. The AR gene was the only overexpressed gene common to all CRCaP xenografts when the expression profiles of all 7 pairs were compared. In addition, AR overexpression converted bicalutamide, flutamide, and cyproterone acetate from AR antagonists to agonists, which suggests that AR overexpression can cause, at least in part, the antiandrogen withdrawal syndrome. Abundance of AR increases the total AR content of the tumor that is available for ligand binding and allows seemingly androgen-independent cancer cells to proliferate in an androgen-depleted environment.
Palmberg and colleagues suggested better response to antiandrogen therapy addition to gonadotropin-releasing hormone analogues in CRCaP patients whose AR was amplified compared with patients without it, which suggested continued dependence of CRCaP on residual androgens. One mechanism of increasing AR expression is through endothelin-1 (ET-1). ET-1 is highly expressed by CaP cell lines and tumor specimens, and elevated plasma levels of ET-1 are present in men with CRCaP. ET-1 binds endothelin A receptor, which activates Src/phosphatidylinositol 3-kinase (PI3K) signaling pathways (see section on the outlaw pathway) and augments AR expression via c-Myc. ET-1 also stimulates osteoblasts and plays a role in osteoblastic bone metastasis.
Increased AR Sensitivity to Androgens
The authors’ group was the first to demonstrate increased stability of AR in the “absence of androgens” when androgen-sensitive LNCaP cells were compared with androgen-independent LNCaP-C4-2 and CWR-R1 cells, and castration-recurrent CWR22 xenograft tumors. AR in LNCaP cells was unstable, with a degradation half-time (t 1/2 ) of 3 hours. By contrast, AR was more stable in androgen-independent CWR-R1 (t 1/2 = 6 hours) and LNCaP-C4-2 cell lines (t 1/2 = 7 hours), and castration-recurrent CWR22 tumors (t 1/2 >12 hours). The concentration of dihydrotestosterone (DHT) required for growth stimulation of androgen-independent cells (CWR-R1 and LNCaP-C4-2) was 4 orders of magnitude lower than that required for LNCaP cells. The increased sensitivity to androgens was not associated with major differences in AR expression, AR binding affinity to androgens, or AR-androgen dissociation times. These results suggested that increased AR stability may increase AR nuclear retention time, which could result in an increased biological response at lower DHT levels with little change in AR levels or affinity. The authors’ group provided a molecular mechanism to explain increased AR sensitivity to low androgen levels in CRCaP; the AR coactivator profile changed from SRC1 to TIF2 in castration-recurrent CWR22 xenografts and CRCaP clinical samples. Other common mechanisms that hypersensitize AR to low androgen levels in CRCaP include growth factor and/or cytokine-induced AR phosphorylation via signal transduction pathways, for example, AR tyrosine phosphorylation at Y534 by Src kinase, and Y267 and Y363 by Ack1 kinase (see section on the outlaw pathway).
Intracrine synthesis of androgens by CRCaP tissue
CRCaP increases the availability of testicular androgen to AR by de novo synthesis of testicular androgens from ubiquitous substrates.
Testosterone (T) is the main circulating testicular androgen, whereas DHT is the main intracellular testicular androgen. DHT is the preferred AR ligand because DHT has higher affinity for AR than T owing to slower off time, and 10-fold higher potency of inducing AR signaling than T. T is converted to DHT by the enzyme 5α-reductase (5α-R). Whereas the DHT:T ratio in benign prostate and ASCaP tissues is 2.5:1 to 20:1, DHT levels drop significantly in CRCaP tissues and the ratio becomes 0.25:1 to 0.5:1. This change in ratios is likely a consequence of changes in 5α-R isozyme tissue expression during CaP progression. The authors’ group and others reported relative increases in 5α-R isozymes 1 and 3, and a relative decrease in isozyme 2 in CRCaP, versus ASCaP. The total 5α-reducing capability in CRCaP versus ASCaP is probably decreased because isozyme 2 is the most efficient 5α-reducing enzyme at physiologic (intracellular) pH. Despite the significant reduction in DHT levels in CRCaP tissues, AR signaling remains active. Comparing levels of T and DHT between benign prostatic and ASCaP tissues on one hand, with CRCaP tissues reveals persistent and even elevated androgen levels in CRCaP tissues, well within the range that is capable of activating wild-type and mutated ARs.
The authors’ group compared prostatic tissue levels of T and DHT between androgen-stimulated benign prostate and CRCaP. T levels were similar and DHT levels were 80% to 90% lower. Mean DHT levels were 8.13 pmol/g versus 1.45 pmol/g tissue using mass spectrometry (MS), and 13.7 pmol/g versus 1.25 pmol/g tissue using radioimmunoassay (RIA), levels sufficient to activate even a molecularly “normal” AR (wild-type unphosphorylated AR with normal coregulator profile). Continued AR activation in CRCaP was suggested by continued expression of PSA. These results were confirmed by others. T level was 3-fold higher, whereas DHT level was 11-fold lower (mean 0.25 ng/g tissue), in metastatic CRCaP lesions compared with prostatic tissue from eugonadal patients with ASCaP. Expression of AR and PSA proteins was higher and similar, respectively, between metastatic CRCaP (bone metastasis) and ASCaP tissues, again indicating continued AR signaling in CRCaP.
Tissue levels of androgens are difficult to measure using either MS or RIA because of problems with tissue procurement, low tissue androgen levels, and assay sensitivity. However, tissue androgen levels appear similar when measured by different groups using different methods to assay different types of tissues ( Table 2 ). In CWR-R1 and LNCaP-C4-2 cell lines (both exhibiting mutated ARs), a DHT level as low as 10 −14 M (2.92 × 10 −6 ng/g tissue) was sufficient for growth stimulation.
| Mass Spectrometry | Radioimmunoassay | ||||
|---|---|---|---|---|---|
| T | DHT | T | DHT | ||
| Titus et al, 2005 | Mohler et al, 2004 | ||||
| ASBP (n = 18) | 2.75 | 13.7 | ASBP (n = 30) | 3.26 | 8.13 |
| CRCaP (n = 18) | 3.75 | 1.25 | CRCaP (n = 15) | 2.78 | 1.45 |
| Montgomery et al, 2008 | Geller et al, 1979 | ||||
| ASBP (n = 6) | 0.04 | 1.92 | ASBP (n = 17) | NM | 17.6 |
| ASCaP (n = 4) | 0.23 | 2.75 | CaP orch ± DES (n = 9) | NM | 4.47 |
| CR-Met CaP (n = 8) | 0.74 | 0.25 | CaP DES 1 mg (n = 6) | NM | 12.4 |
| Labrie et al, 1989 | |||||
| Human CaP (n = NS) | NM | 18.6 | |||
| Orch (n = 5, 2–12 mo) | NM | 9.29 | |||
| Orch + FL (n = 4, 2 mo) | NM | ND | |||
Substrates for de novo androgen synthesis in CRCaP cells include cholesterol, cholesterol precursors (eg, acetic acid), and adrenal androgens. Furthermore, all enzymes necessary for DHT synthesis from cholesterol are expressed, and most are upregulated (3–30 fold) in CRCaP tissue.
Two closely intertwined pathways exist that ultimately lead to DHT synthesis from cholesterol: the front-door pathway (via the adrenal androgens dehydroepiandrosterone [DHEA] and androstenedione [ASD]) and the back-door pathway (via pregnanedione and androstanediol) ( Fig. 2 ). In the classic pathway, C21 steroids such as pregnenolone and progesterone are first converted to the C19 adrenal androgens DHEA and ASD via sequential 17α-hydroxylase and 17,20-lyase activities of CYP17A, followed by the activities of 17β-hydroxysteroid dehydrogenase (17βHSD) and 5α-R. In the back-door pathway, the order of steps is reversed whereby C21 steroids are first 5α-reduced, followed by sequential CYP17A and 17βHSD actions. Androstanediol is the major degradation product of DHT from the reductive 3α-hydroxysteroid dehydrogenase (HSD) activity of 3α-HSD aldo-keto reductases 1C (AKR1C). The authors’ group demonstrated AR transactivation by androstanediol in benign and malignant prostate-derived cell lines via oxidation of androstanediol into DHT. Administration of androstanediol to castration-recurrent CWR22 tumor xenografts resulted in a 28-fold increase in intratumoral DHT levels.
Clinical indicators of the significance of residual prostatic tissue androgens in CRCaP include the benefits of secondary or tertiary hormonal therapies in CRCaP patients. Ketoconazole administration in CRCaP is associated a PSA response (≥50% decrease in PSA from baseline) in 50% to 60% of patients. 5α-Reductase inhibitors (5α-RI) used singly or in combination with an antiandrogen or ketoconazole and hydrocortisone resulted in PSA decreases of variable magnitudes and durations in more than half of the patients with CRCaP. Abiraterone, a CYP17A inhibitor of adrenal and testicular androgen synthesis, prolongs overall survival in post-docetaxel CRCaP patients by nearly 4 months. Abiraterone also decreases “castrate” serum T level in CRCaP patients from less than 50 ng/dL to less than 1 ng/dL.
These results strongly support that CRCaP tissue is capable of intracrine testicular androgen synthesis from ubiquitous substrates. Therapeutic strategies designed to more effectively ablate tumor androgens are needed and are likely to improve CRCaP outcomes.
The promiscuous pathway
AR specificity is broadened such that AR could be activated by nonandrogenic steroids or even antiandrogens. The broadened specificity might be achieved either through mutations in the LBD or through alterations in the coregulator profile.
AR Mutations
The AR gene is not necessary for survival, as is evident by the germline loss-of-function mutations that lead to androgen insensitivity syndrome. CRCaP that acquires somatic gain-of-function mutations in the LBD of the AR gene have broadened ligand specificity, which allows AR activation by nontesticular androgens and antiandrogens. AR mutations seen in androgen insensitivity syndrome and CRCaP involve different parts of the AR gene. AR mutations can explain the antiandrogen withdrawal syndrome observed on discontinuation of antiandrogens (flutamide, nilutamide, bicalutamide, and cyproterone acetate ).
The first such AR mutation was identified in LNCaP cells, which were activated by testicular and adrenal androgens, progesterone, estradiol, and flutamide, but not bicalutamide. Sequencing the AR gene from LNCaP cells revealed a missense mutation in amino acid 877, located in the LBD. This mutation caused the substitution of alanine for threonine (T877A). CaP cells possessing this mutation were able to be clonally selected to grow in a castrate environment, leading to CRCaP.
The frequency of AR mutations reported in the literature varies widely (0%–50%). AR mutations were initially reported in 50% (5 of 10) of patients with CRCaP from whom specimens were obtained from metastatic lesions (bone marrow, skin, and pleural fluid) using a cloning-based method whereby polymerase chain reaction integrity may have been problematic. Subsequent reports from the same group identified AR mutations at lower frequencies. AR mutations were reported in 10% to 30% of CRCaP patients who were treated for long periods of time with castration and antiandrogens, and specimens were obtained from bone marrow metastases. Work by the authors’ group using different methods suggested similar frequency of AR mutations in CRCaP. The T877A mutation was detected in 10% of 25 prostatectomy specimens from CRCaP patients using denaturing gradient gel electrophoresis (DGGE) of exons B to H, and direct sequencing of exon A and any possible mutations suggested by DGGE. Factors that make it difficult to determine the true in vivo frequency of AR mutations include patient selection, tumor heterogeneity, tissue source (prostate gland vs metastases), method of tissue preservation, duration of antiandrogen therapy, and molecular methods.
The human CaP xenograft CWR22 cells have an H874Y (substitution of tyrosine for histidine) mutant AR that is activated by testicular and adrenal androgens, estradiol, progesterone, and flutamide. The H874Y mutation occurs in the LBD and broadens AR specificity by changing coactivator binding to AR. Passage of these cells in castrate nude mice gives rise to castration-recurrent CWR22 cells, also known as CWR-R1 cells, which exhibit the same AR mutation.
MDA PCa 2a and 2b cell lines were established from bone metastasis in a CRCaP patient. These cell lines harbor a promiscuous AR that has reduced affinity for androgens. Two distinct missense mutations in the AR LBD were detected in these cell lines: the T877A mutation and the leucine-to-histidine substitution at amino acid 701 (L701H). The L701H mutation alone decreases AR affinity for DHT and increases affinity to cortisol. The T877A mutation broadens AR specificity to include progesterone and estradiol, and provides a synergistic effect by increasing AR affinity for cortisol 3-fold more than the L701H mutation alone. Cortisol was associated with the highest transactivation response in L701H-T877A transfected cells. The frequency of this mutation in CRCaP remains unknown.
Recent work that reported AR transactivation by androstanediol demonstrated the ability of CRCaP cells to convert a weak androgen into DHT. Other weak AR ligands may be converted to testicular androgens by CRCaP cells instead of transactivating a promiscuous AR directly. This new concept questions the significance of AR mutations and must cause a reexamination of publications describing broadened specificity of any steroid receptor for its ligands.
Many other mutations in the AR gene have been identified and are cataloged in the Androgen Receptor Gene Mutations Database ( http://androgendb.mcgill.ca/ ), but it is unclear how many other mutations use the promiscuous receptor mechanism.
Altered Coregulator Profiles
Several proteins modulate the function of AR and act as either coactivators or corepressors of DNA transcription. More than 170 AR coregulator proteins have been reported. Each coregulator either interacts directly with AR or is found in a transcriptional complex that includes AR. Alterations in AR coregulator levels and function may contribute to the development of CRCaP. In addition, overexpression of some coregulators in CaP clinical specimens was associated with unfavorable clinical outcomes.
Increase in coactivator proteins
Several AR coactivators, such as TIF2, SRC1, Tip60, and CARM1, are overexpressed in CRCaP. Proposed mechanisms for AR coactivator–induced growth stimulation in CRCaP include increased AR sensitivity to ligands and/or through broadened AR specificity.
Clinical samples of androgen-stimulated benign prostate, ASCaP, CRCaP, and androgen-stimulated and castration-recurrent CWR22 tumor xenografts were used to detect overexpression of AR and 2 AR coactivators; TIF2 and SRC1, in CRCaP samples. TIF2 was more overexpressed than SRC1 so that the transcriptional complex had a predominance of TIF2. Overexpression of TIF2 in cotransfection assays increased AR transcriptional activity in the presence of physiologic concentrations of adrenal androgens. Furthermore, overexpressed TIF2 increased the transcriptional activity of the mutant ARs (T877A in LNCaP) and (H874Y in CWR22) in the presence of unchanged levels of testicular and adrenal androgens, progesterone, estradiol, and hydroxyflutamide. These results show that overexpression of TIF2 and/or SRC1 in CRCaP increases AR transactivation in wild-type and mutant ARs in response to low and physiologic concentrations of testicular and adrenal androgens, and other steroids with affinity for AR. Agoulnik and colleagues found that reducing SRC1 expression decreased cell growth and PSA synthesis in the AR-dependent LNCaP and LNCaP-C4-2 cell lines, whereas proliferation was unaffected in the AR-negative PC-3 and DU145 cell lines, suggesting that the effects of SRC1 are AR-dependent. Unlike SRC1, the effects of TIF2 seem to be a combination of AR-dependent and AR-independent mechanisms, because reducing the expression of TIF2 negatively affected the growth of both LNCaP and PC-3 cells.
Yeh and colleagues transfected DU145 CaP cells with AR and found that overexpression of the AR coactivator (ARA70) enhanced AR transactivation 10-fold in the presence of DHT or T, and greater than 30-fold in the presence of estradiol.
Halkidou and colleagues studied the expression and subcellular localization of the AR coactivator Tip60 in CaP cell lines, and in benign and malignant prostate clinical samples. On androgen withdrawal, Tip60 expression was upregulated and shifted to the nucleus in LNCaP cell culture and in CWR22 xenografts. Androgen exposure decreased the expression of Tip60 and increased its cytoplasmic distribution. In clinical samples, Tip60 had a diffuse cellular distribution in benign prostatic and ASCaP tissues, whereas in CRCaP, localization was mainly nuclear.
The AR coactivator CARM1 is overexpressed in CRCaP but not in ASCaP. Reducing CARM1 expression decreased PSA expression and CaP cell growth, and induced apoptosis. Many other coactivators have been identified, but their contributions to AR action have not been characterized fully.
Decrease in corepressor proteins
Corepressors reduce AR activity through a variety of mechanisms such as prevention of nuclear localization and DNA binding, recruiting histone deacetylases that deacetylate chromatin and other proteins required for transcriptional activity, competing with coactivators for binding to AR, reducing NTD/LBD interactions required for optimal transcriptional activation of AR, and functioning as scaffolds for other AR coregulators. Examples include NCoR1, SMRT/NCoR2, DAX-1, cyclin D1a (an estrogen-receptor coactivator), histone deacetylases, and the less well characterized proteins Hey1 and FoxH1. Recruitment of NCoR1 and SMRT by agonist-bound AR has been shown to suppress agonist-induced activation of androgen-regulated genes such as PSA, TSC22, NKX3.1 and B2M. Recruitment of corepressors is believed to be a key mediator of antagonist-mediated inhibition of steroid receptors including AR, which suggests that loss of expression of corepressors could facilitate tumor growth in the presence of AR antagonists used in conjunction with castration. The large number of AR coregulators and their inherent redundancy make AR coregulators challenging targets for drug development.
Related posts:
 Moving Toward Personalized Medicine in Castration-Resistant Prostate Cancer
Moving Toward Personalized Medicine in Castration-Resistant Prostate Cancer
 Palliative Care in Castrate-Resistant Prostate Cancer
Quality of Life with Advanced Metastatic Prostate Cancer
The Experience with Cytotoxic Chemotherapy in Metastatic Castration-Resistant Prostate Cancer
Palliative Care in Castrate-Resistant Prostate Cancer
Quality of Life with Advanced Metastatic Prostate Cancer
The Experience with Cytotoxic Chemotherapy in Metastatic Castration-Resistant Prostate Cancer
 Management of Docetaxel Failures in Metastatic Castrate-Resistant Prostate Cancer
Management of Docetaxel Failures in Metastatic Castrate-Resistant Prostate Cancer
 Targeting Angiogenesis as a Promising Modality for the Treatment of Prostate Cancer
Targeting Angiogenesis as a Promising Modality for the Treatment of Prostate Cancer
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


