CHAPTER 88 Autoimmune Hepatitis
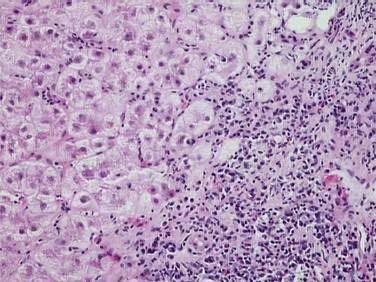
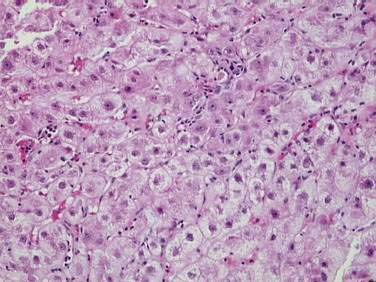
Autoimmune hepatitis (AIH) is a disorder of unknown cause characterized by unresolving inflammation of the liver and by the presence of interface hepatitis on histologic examination (Fig. 88-1), hypergammaglobulinemia, and autoantibodies.1 Diagnosis requires the exclusion of other chronic liver diseases that have similar features, including Wilson disease, chronic viral hepatitis, drug-induced liver disease, nonalcoholic fatty liver disease, and the immune cholangiopathies of primary biliary cirrhosis (PBC) and primary sclerosing cholangitis (PSC). Panacinar or lobular hepatitis is within the histologic spectrum of AIH (Fig. 88-2), and centrilobular (Rappaport zone 3) necrosis has been described in AIH and may indicate an early stage of the disease before the development of interface hepatitis.2
DIAGNOSTIC CRITERIA
An international panel codified the diagnostic criteria of AIH in 1992, and an expanded panel updated them in 1999.3 The propensity for an acute, rarely fulminant, presentation has been recognized,2 and the requirement for six months of disease activity to establish chronicity has been waived.3 Cholestatic histologic changes, including bile duct injury and ductopenia, are incompatible findings, but trivial biliary changes within the background of classic histologic features do not preclude the diagnosis.4,5
The serologic tests essential for diagnosis are assays for antinuclear antibodies (ANA), smooth muscle antibodies (SMA), and antibodies to liver-kidney microsome type 1 (anti-LKM1).1,6,7 These assays are based on the indirect immunofluorescence of rodent tissues or Hep-2 cell lines or on enzyme immunoassays using microtiter plates with adsorbed recombinant or highly purified antigens. Atypical perinuclear anti-neutrophil cytoplasmic antibodies (atypical pANCA) are common in type 1 AIH, PSC, and chronic ulcerative colitis.6,7 They are directed against antigens within the nucleus, rather than the cytoplasm, of granulocytes, and the reactivities localize to the proteins within the lamina of the nucleus. Because these antibodies are directed against nuclear envelope antigens rather than cytoplasmic antigens, a better term for them is anti-neutrophil nuclear antigens or “ANNA.”8 Atypical perinuclear anti-neutrophil cytoplasmic antibodies have been useful in evaluating patients who lack the conventional autoantibodies.6,7 Celiac disease can be associated with liver disease that resembles AIH and should be excluded in patients with cryptogenic chronic hepatitis by screening for immunoglobulin (Ig) A antibodies to tissue transglutaminase or endomysium.6,7,9 IgA endomysial antibodies are more predictive of celiac disease in patients with AIH than are IgA antibodies to tissue transglutaminase, which can be stimulated by hepatic inflammation and fibrogenesis.6,7
New autoantibodies continue to be characterized in the hope of improving diagnostic specificity and prognostic value, but none has been incorporated into conventional diagnostic algorithms.6,7,10 Antibodies to soluble liver antigen (anti-SLA), actin (anti-actin), chromatin (anti-chromatin), asialoglycoprotein receptor (ASGPR), and liver cytosol type 1 (anti-LC1) have been associated with severe AIH, poor treatment response, and relapse after drug withdrawal.10–13 Their major clinical limitation has been their infrequent individual occurrence in AIH.
CLINICAL CRITERIA
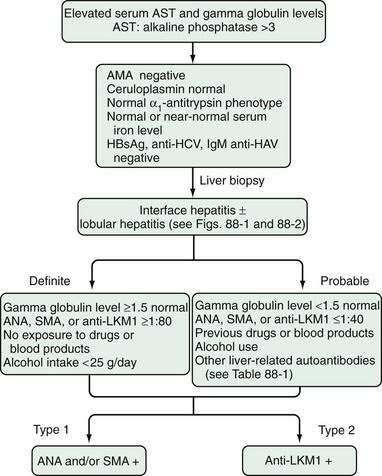
The definite diagnosis of AIH requires exclusion of other similar diseases; laboratory findings that indicate substantial immunoreactivity; and histologic features of interface hepatitis (Fig. 88-3).3 A probable diagnosis is justified when findings are compatible with AIH but insufficient for a definite diagnosis (see Fig. 88-3).3 Patients who lack conventional autoantibodies but who are seropositive for investigational markers, such as antibodies to ASGPR, SLA, actin, or LC1, are classified as having probable disease.3
SCORING CRITERIA
The original scoring system proposed by the International Autoimmune Hepatitis Group accommodates the diverse manifestations of AIH and renders an aggregate score that reflects the net strength of the diagnosis before and after glucocorticoid treatment (Table 88-1).3 By weighing each component of the syndrome, discrepant features can be accommodated and biases associated with isolated inconsistencies can be avoided. The original scoring system ensures the comparability of study populations in clinical trials, provides a comprehensive template for systematically assessing all features of the disease, and can render a diagnosis of AIH in patients with few or atypical features.3 The original scoring system is not a discriminative diagnostic index, and it should not be used to distinguish AIH from other liver diseases.
Table 88-1 Revised Original Scoring System for the Diagnosis of Autoimmune Hepatitis
| CATEGORY | VARIABLE | SCORE |
|---|---|---|
| Gender | Female | +2 |
| AP/AST | >3 | −2 |
| <1.5 | +2 | |
| Gamma globulin or IgG level above normal | >2.0* | +3 |
| 1.5-2.0* | +2 | |
| 1.0-1.5* | +1 | |
| <1.0 | 0 | |
| ANA, SMA, or anti-LKM1 titer | >1 : 80 | +3 |
| 1 : 80 | +2 | |
| 1 : 40 | +1 | |
| <1 : 40 | 0 | |
| AMA | Positive | −4 |
| Viral markers | Positive | −3 |
| Negative | +3 | |
| Drug history | Yes | −4 |
| No | +1 | |
| Alcohol | <25 g/day | +2 |
| >60 g/day | −2 | |
| HLA | DR3 or DR4 | +1 |
| Immune disease | Thyroiditis, ulcerative colitis, synovitis, others | +2 |
| Other liver-defined autoantibodies | Anti-SLA, anti-actin, anti-LC1, pANCA | +2 |
| Histologic features | Interface hepatitis | +3 |
| Plasmacytic infiltrate | +1 | |
| Rosettes | +1 | |
| None of above | −5 | |
| Biliary changes | −3 | |
| Other features | −3 | |
| Treatment response | Complete | +2 |
| Relapse | +3 | |
| Pretreatment Score | ||
| Definite diagnosis | >15 | |
| Probable diagnosis | 10-15 | |
| Post-treatment Score | ||
| Definite diagnosis | >17 | |
| Probable diagnosis | 12-17 |
AMA, antimitochondrial antibodies; ANA, antinuclear antibodies; anti-LC1, antibodies to liver cytosol type 1; anti-LKM1, antibodies to liver-kidney microsome type 1; anti-SLA, antibodies to soluble liver antigen; AP/AST (or AP/ALT), ratio of serum alkaline phosphatase level to serum aspartate aminotransferase (or serum alanine aminotransferase) level; HLA, human leukocyte antigen; IgG, immunoglobulin G; pANCA, perinuclear anti-neutrophil cytoplasmic antibodies; SMA, smooth muscle antibodies.
* Times upper limit of normal.
Adapted from Alvarez F, Berg PA, Bianchi FB, et al. International Autoimmune Hepatitis Group report: Review of criteria for diagnosis of autoimmune hepatitis. J Hepatol 1999; 31:929-38.
A simplified scoring system has been developed to ease clinical application and is based on four clinical components that include the presence and level of autoantibody expression by indirect immunofluorescence, serum IgG concentration, histologic features, and viral markers (Table 88-2).14 The original scoring system has greater sensitivity for the diagnosis than the simplified system (100% vs. 95%), but the simplified system has greater specificity (90% vs. 73%) and predictability (92% vs. 82%).15 Whereas the original scoring system is useful for evaluating patients in whom every component must be assessed because of few or atypical findings, the simplified scoring system is useful for excluding AIH in patients with other conditions and concurrent immune features.
Table 88-2 Simplified Scoring System for Diagnosis of Autoimmune Hepatitis*
| CATEGORY | VARIABLE | SCORE |
|---|---|---|
| Autoantibodies† | ||
| Antinuclear antibodies or smooth muscle antibodies | 1 : 40 | +1 |
 1 : 80 1 : 80 | +2 | |
| Antibodies to liver-kidney microsome type 1 | 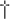 1 : 40 1 : 40 | +2 |
| Antibodies to soluble liver antigen | Positive | +2 |
| Immunoglobulin Level | ||
| Immunoglobulin G | >Upper limit of normal | +1 |
| >1.1 times upper limit of normal | +2 | |
| Histologic Findings | ||
| Morphologic features | Compatible with autoimmune hepatitis | +1 |
| Typical of autoimmune hepatitis | +2 | |
| Viral Disease | ||
| Absence of viral hepatitis | No viral markers | +2 |
| Pretreatment Aggregate Score | ||
| Definite diagnosis | ≥7 | |
| Probable diagnosis | 6 |
* Adapted from Hennes EM, Zeniya M, Czaja AJ, et al. Simplified diagnostic criteria for autoimmune hepatitis. Hepatology 2008; 48:169-76.
† Autoantibody titers as determined by indirect immunofluorescence.
PATHOGENESIS
The pathogenic mechanisms of AIH are unknown. The most popular hypotheses invoke a constellation of interactive factors that include a triggering agent, genetic predisposition, and various determinants of autoantigen display, immunocyte activation, and effector cell expansion (Fig. 88-4).16–18 Proposed triggering factors include infectious agents, drugs, and toxins. The lag time between exposure to the trigger and onset of the disease can be long, and the triggering factor may not be needed for perpetuation of the disorder. The CD4+ helper T cell is the principal effector cell, and its activation is the initial step in the pathogenic pathway.
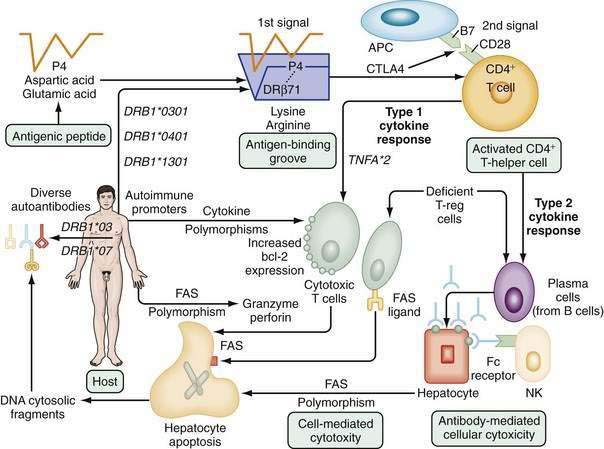
Molecular mimicry of a foreign antigen and a self-antigen is the most common explanation for the loss of self-tolerance, but this mechanism has not been established for any autoimmune disease.16–18 Genetic factors influence autoantigen presentation and CD4+ helper T cell recognition. The antigen-binding groove of the class II molecule of the major histocompatibility complex (MHC) is encoded by alleles that determine the groove’s configuration and ability to activate immunocytes. The susceptibility alleles of AIH in white North Americans and northern Europeans reside on the DRB1 gene and are DRB1*0301 and DRB1*0401 (see Fig. 88-4).19–21
Different ethnic groups have different susceptibility alleles, a finding that supports a “shared motif hypothesis” of pathogenesis.19–21 According to this hypothesis, the risk of disease relates to amino acid sequences in the antigen-binding groove of the class II MHC molecule, and multiple alleles encode the same or similar sequence (“shared motif”). The critical shared motif in white North Americans and northern Europeans with AIH is a six-amino-acid sequence represented by the code LLEQKR.19–21 This sequence is located between positions 67 and 72 of the DRβ polypeptide chain of the class II MHC molecule, and lysine (K) in position 71 is the critical determinant of susceptibility. DRB1*0301 and DRB1*0401 encode identical amino acid sequences in the DRβ67-72 region and affect susceptibility similarly.
DRB1*0404 and DRB1*0405 are the susceptibility alleles in Mexican, Japanese, mainland Chinese, and Argentine adults and encode a similar sequence, except for arginine (R) instead of lysine (K) at the DRβ71 position.20,21 Arginine is a positively charged amino acid that is structurally similar to lysine, and its substitution for lysine should not greatly alter the antigen-binding properties of the class II MHC molecule. By contrast, DRB1*1501 protects against AIH in white North Americans and northern Europeans, and this allele encodes isoleucine (I) instead of leucine (L) at position DRβ67 and alanine (A) instead of lysine (K) at position DRβ71. Alanine is a neutral, nonpolar amino acid that, when substituted for lysine, should greatly affect antigen presentation and immunocyte activation.
Antigenic peptides are selected for display by the nature of the amino acids that interact with residues within the antigen-binding groove (see Fig. 88-4).21,22 The critical six-amino-acid motif in AIH restricts the range of peptides that can be accommodated. Multiple self-antigens or foreign antigens may satisfy the minimal structural requirements and serve as immunogenic peptides. The ideal triggering epitope must have a negatively charged amino acid residue (aspartic acid or glutamic acid) at peptide position P4 to form a salt bridge with the positively charged lysine or arginine at DRβ71.21 Molecular modeling indicates that a negatively charged P4 residue in the antigenic peptide and the positively charged lysine or arginine at DRβ71 can form a P4-DRβ71 immunoreactive unit that is independent of the other residues within the antigen and antigen-binding groove. This minimal immunoreactive unit can be created by multiple antigenic peptides and class II MHC molecules, and the number of these units may affect susceptibility by a “dose effect.”
DRB1*1301 is associated with AIH in Argentine children22 and Brazilian patients23,24 and encodes ILEDER at positions DRβ67-72. Glutamic acid (E), aspartic acid (D), and glutamic acid (E) are at positions DRβ69, DRβ70, and DRβ71, respectively, in the class II MHC molecule, and the presence of these critically located but negatively charged amino acid residues argues against the “shared motif” hypothesis of pathogenesis. The “molecular footprint” hypothesis of pathogenesis holds that susceptibility to AIH in different regions and ethnic groups relates to indigenous factors or agents favored by certain genetic phenotypes.20,21 In South America, DRB1*1301 is associated with protracted hepatitis A virus infection, and persons with this allele may be “selected” from their environment to have prolonged exposure to viral and hepatic antigens that favor the development of AIH.25 An understanding of the individual susceptibility allele in different geographic regions may allow use of this “footprint” to track the cause of the disease.
The “autoimmune promoter hypothesis” of pathogenesis complements the “shared motif” and “molecular footprint” hypotheses by proposing that genetic promoters inside and outside the MHC can affect disease occurrence, either in synergy (epistasis) with the principal susceptibility factors or in lieu of them.18,20,21 Polymorphisms of the tumor necrosis factor (TNF)-α gene (TNFA*2),26 the cytotoxic T lymphocyte antigen 4 gene (CTLA4),27 and Fas gene promoter at position −670 (TNFRSF6)28 have been associated with increased immunoreactivity, disease severity, and early progression to cirrhosis in white North American and northern European patients. Constellations of autoimmune promoters, as yet undefined, may individualize the disease by affecting its occurrence, clinical phenotype, and outcome.
Liver cell destruction is accomplished by either cell-mediated cytotoxicity, antibody-dependent cell-mediated cytotoxicity, or a combination of both mechanisms (see Fig. 88-4).16–18 Cell-mediated cytotoxicity depends on the clonal expansion of CD8+ cytotoxic T cells that accomplish liver cell injury through the release of lymphokines. This mechanism is regulated by type 1 cytokines, and the −308 polymorphism of TNFA*2 may facilitate this pathway.26 Antibody-dependent cell-mediated cytotoxicity is regulated by type 2 cytokines, and the natural killer cell accomplishes liver cell destruction by binding of its Fc receptor with an antigen-antibody complex on the hepatocyte surface.16–18 The predominant mechanism depends on the phenotypic differentiation of the CD4+ helper T cell, which in turn reflects the cytokine milieu. The cytokine milieu may reflect polymorphisms of the cytokine genes that favor excessive production of some modulators, such as TNF-α, or deficient production of others.
Defects in the counter-regulatory cytokine milieu may also reflect reduced numbers of intrahepatic natural killer T (NKT) cells and the failure of T-regulatory (T-reg) cells (CD4+CD25+ cells) to modulate CD8+ T cell proliferation and cytokine production.18,29 Increased numbers of γδ T cells may also contribute to the cytodestructive process by recognizing antigens presented by the non-classic MHC molecules, and the recruitment and intrahepatic trafficking of cytotoxic T lymphocytes may be enhanced by the up-regulation of chemokines, such as CXCL16.18 Fibrogenesis, in turn, is fueled by the resulting inflammatory activity as perivascular hepatic stellate cells transform into myofibroblasts. The matrix proteins accumulate as tissue inhibitors retard the counteractive degradative actions of matrix metalloproteinases, and stellate cells continue to be activated in an autocrine fashion by transforming growth factor-β (TGF-β).30 Glucocorticoid therapy can favorably alter the cytokine milieu, improve the number and function of the T-reg cells, impair activation of TGF-β, promote disappearance of the metalloproteinase inhibitors, enhance degradation of the fibrotic liver matrix, and foster apoptosis of the hepatic stellate cells (see also Chapter 2).31
CLASSIFICATION
Two types of AIH have distinctive serologic profiles. Neither has been ascribed a unique cause, specific treatment strategy, or special type of behavior (Table 88-3). The terms are useful as clinical descriptors and as research designations to ensure homogeneous study populations.
Table 88-3 Classification of Autoimmune Hepatitis Based on Autoantibodies
| CLINICAL FEATURE | TYPE 1 | TYPE 2 |
|---|---|---|
| Signature autoantibodies | Smooth muscle | Liver-kidney microsome type 1 |
| Nuclear | ||
| Associated autoantibodies | Atypical pANCA | Liver cytosol type 1* |
| Actin* | Soluble liver antigen* | |
| Asialoglycoprotein receptor* | ||
| Chromatin* | ||
| Soluble liver antigen* | ||
| Putative autoantigen | Unknown | CYP2D6 |
| Age | Infants to elderly | Children (2-14) |
| Female | 78% | 89% |
| Concurrent immune diseases | 38% | 34% |
| Typical concurrent autoimmune diseases | Autoimmune thyroiditis | Autoimmune thyroiditis |
| Graves’ disease | Vitiligo | |
| Ulcerative colitis | Type 1 diabetes mellitus | |
| APECED | ||
| Organ-specific antibodies | 4% | 30% |
| Serum gamma globulin elevation | +++ | + |
| HLA associations | B8, DR3, DR4, DR13 | B14, DR3, C4A-Q0, DR7 |
| Allelic risk factors | DRB1*0301 and *0401 | DQB1*0201, DRB1*07, DRB1*03 |
| (white North Americans and northern Europeans) | (Germans and Brazilians) | |
| DRB1*1301 | ||
| (South Americans, especially children) | ||
| Glucocorticoid responsive | +++ | ++ |
APECED, autoimmune polyendocrinopathy–candidiasis–ectodermal dystrophy; CYP2D6, cytochrome P450 2D6; pANCA, perinuclear anti-neutrophil cytoplasmic antibodies.
* Autoantibodies with an asterisk are investigational only and not available for routine clinical use.
TYPE 1 AUTOIMMUNE HEPATITIS
Type 1 AIH is characterized by SMA, ANA, or both (see Table 88-3).1,7 Antibodies to actin have greater specificity, but less sensitivity, for the diagnosis of AIH than SMA.6 Atypical pANCA are found in as many as 90% of patients with type 1 AIH and typically are absent in type 2 AIH.6,7
Type 1 AIH can occur at any age and in either gender (see Table 88-3).7 Initial studies that suggested a bimodal age distribution probably reflected referral biases to tertiary medical centers. The disease has been described in infants and probably is underdiagnosed in the elderly.32 Seventy-eight percent of patients are women (female-to-male ratio 3.6 : 1), and 38% have concurrent extrahepatic immunologic diseases.7 Autoimmune thyroiditis (occurring in 12% of the cases), Graves’ disease (6%), and ulcerative colitis (6%) are the most common associated immune disorders. Rheumatoid arthritis, pernicious anemia, scleroderma, Coombs-positive hemolytic anemia, autoimmune thrombocytopenic purpura, symptomatic cryoglobulinemia, leukocytoclastic vasculitis, nephritis, erythema nodosum, systemic lupus erythematosus, and fibrosing alveolitis also may occur (less than 1% each). Cholangiography is warranted to exclude PSC in all patients who have concurrent ulcerative colitis (see Chapter 68).33
Type 1 AIH is associated with an abrupt onset of symptoms in 40% of cases and may manifest in a fulminant fashion.2 The acute presentation frequently reflects preexisting subclinical disease that is unmasked by progression or represents a spontaneous exacerbation of inflammatory activity. Features of chronicity are lacking in 8% of patients, in whom the presentation of the disorder is indistinguishable from that of acute viral or toxic hepatitis.
The target autoantigen of type 1 AIH is unknown. Human leukocyte antigen (HLA)-DR3 (DRB1*0301) and HLA-DR4 (DRB1*0401) are independent risk factors for the disease in white North Americans and northern Europeans.19–21 More than 80% of white patients in Great Britain and the United States possess either DRB1*0301, DRB1*0401, or both, compared with 42% of the unaffected white population. In South America, especially in children, DRB1*1301 is the principal susceptibility allele. These findings indicate that type 1 AIH is a complex polygenic disorder.
TYPE 2 AUTOIMMUNE HEPATITIS
Type 2 AIH is characterized by the expression of anti-LKM1 (see Table 88-3).1,7,34 Most affected persons are children (ages 2 to 14 years), but in Europe, especially in Germany and France, 20% of patients are adults.34 In the United States, type 2 AIH is rare, and only 4% of patients older than 18 years have anti-LKM1.35 The regional differences in prevalence may relate to ethnic differences in the genetic predisposition to the disease.36
Type 2 patients are younger than type 1 patients and may have different clinical and laboratory features (see Table 88-3).1,7,34 As with type 1 AIH, an acute or fulminant presentation is possible and important to recognize and treat early. Susceptibility to type 2 AIH has been associated with DQB1*0201, DRB1*07, and DRB1*03.37 DQB1*0201 is in strong linkage disequilibrium with DRB1*07 and DRB1*03 and has been proposed as the principal genetic determinant of the disease. The expression of anti-LKM1 has been associated with DRB1*07, and various aspects of type 2 AIH may have different genetic determinants.38
The target antigen of type 2 AIH is the cytochrome P450 2D6 mono-oxygenase (CYP2D6).39 This protein is a 50-kd microsomal drug-metabolizing enzyme, and its expression on the hepatocyte surface can be modulated by interleukins and TNF-α. Antibodies to LKM1 inhibit the activity of CYP2D6 in vitro but not in vivo, and lymphocytes extracted from the liver tissue of patients who have the disease exhibit immunoreactivity specific to the antigen.
CYP2D6 has been sequenced, cloned, and mapped, and five epitopes are recognized by anti-LKM1.40
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


