CHAPTER 14
Antibiotics for the therapy of gastrointestinal diseases
Introduction
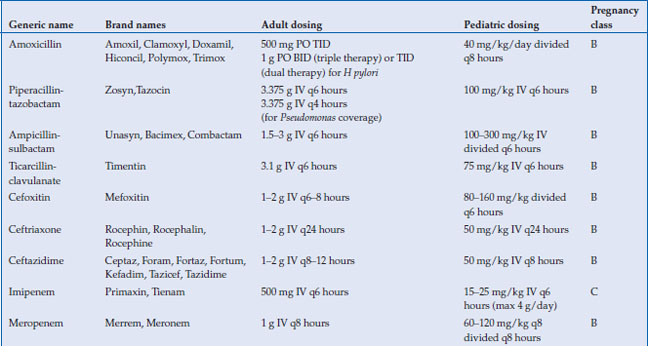
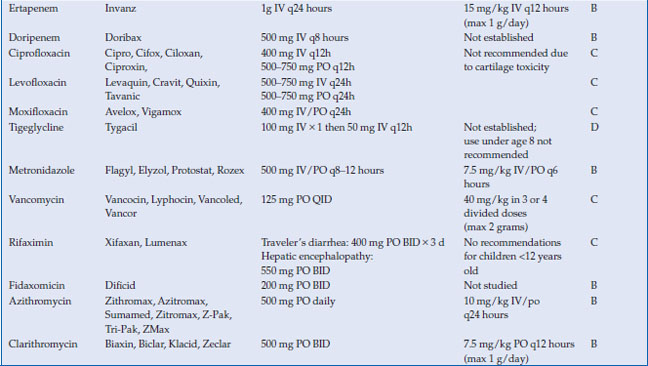
Antimicrobials are used to treat a wide variety of gastrointestinal infections, including infectious diarrhea, Clostridium difficile colitis, Helicobacter pylori and intra-abdominal infections, such as cholecystitis, diverticulitis, appendicitis and abscess. Virtually every class of antimicrobial can be used for some type of infection related to the gastrointestinal system. In this chapter, the most commonly used antimicrobials to treat these infections will be discussed (Table 14.1). The choice of antibiotic depends upon whether the causative agent is known (as with C. difficile or H. pylori) or whether treatment is empiric (as in diverticulitis or cholecystitis). Antivirals for viral hepatitis and antiparasitics for helminths will not be discussed.
Table 14.1 Antimicrobials for the treatment of gastrointestinal infections
Pharmacologic properties
Beta-lactams
The beta-lactams include the penicillins and the cephalosporins. They share a similar ring structure, with side chains determining antibacterial spectrum and pharmacologic properties. Both classes work by inhibiting cell wall synthesis via binding to penicillin-binding proteins (PBPs). Resistance to beta-lactams is conferred by bacterial enzymes that hydrolyze the beta-lactam ring (beta-lactamases), which can be overcome by the addition of a beta-lactamase inhibitor to the penicillin. All beta-lactams are considered bactericidal.
The bioavailability of the penicillins and cephalosporins vary widely, but oral absorption is moderate at best. The most commonly used beta-lactams for gastrointestinal infections are amoxicillin, piperacillin-tazobactam, ampicillin-sulbactam, ticarcillin-clavulanic acid, cefoxitin, ceftriaxone, and ceftazidime. All except amoxicillin are only available intravenously. With the exception of ceftriaxone, all are metabolized by the kidney and thus require dosage adjustment in kidney disease. Ceftriaxone is excreted largely through the biliary system. Both penicillins and cephalosporins are highly protein bound, and they therefore do not penetrate well intracellularly. The distribution into most tissues, however, is adequate. The most important toxicities with the beta-lactams are hypersensitivity reactions, including anaphylaxis, rash, and allergic interstitial nephritis. Cross-reactivity between penicillin allergy and cephalosporin allergy is variable and depends on the nature of the penicillin allergy, as well as the similarity between the side chain of the penicillin causing the reaction and the cephalosporin to be used.
Carbapenems
There are four carbapenems approved for clinical use: imipenem, meropenem, ertapenem, and doripenem. Like the beta-lactams, the carbapenems inhibit bacterial cell wall synthesis by binding to penicillin binding proteins. They are not well-absorbed orally and must therefore be administered intravenously. All are metabolized by the kidney and require dose-adjustment in the presence of renal insufficiency. Drug interactions are minimal, although levels of valproic acid can be lowered by carbapenems. Adverse effects are primarily related to hypersensitivity. All of the carbapenems (imipenem in particular) lower the seizure threshold and have been associated with seizures in clinical use. Importantly, ertapenem does not provide coverage against Pseudomonas aeruginosa, so its use should be avoided in infections where this pathogen is suspected. All other carbapenems possess good coverage against Pseudomonas.
Fluoroquinolones
The fluoroquinolones are synthetic antimicrobials, with each successive generation of agents having a different antimicrobial spectrum. The most widely used fluoroquinolones are the second-generation agents ciprofloxacin and levofloxacin and the fourth generation agent moxifloxacin. Ciprofloxacin and levofloxacin have gram-negative coverage (including susceptible Pseudomonas), with levofloxacin having more gram-positive coverage than ciprofloxacin. Moxifloxacin offers broad spectrum coverage against gram-positive, gram-negative and anaerobic organisms, but not Pseudomonas.
All of the fluoroquinolones act by inhibiting two bacterial enzymes, DNA gyrase and topoisomerase IV. Through inhibition of these enzymes, bacterial cell replication is impaired, followed rapidly by cell death. They are well absorbed and penetrate into tissues well, with levels in some tissues exceeding serum levels. The bioavailability of ciprofloxacin is ~70–80%, and even higher for levofloxacin (99%) and moxifloxacin (90%). Therefore, oral therapy is nearly equivalent to parenteral therapy in persons with an intact small bowel. Once absorbed, ciprofloxacin and levofloxacin are metabolized by the kidney (and require dose adjustment in renal disease), while moxifloxacin is metabolized by the hepatic and biliary systems.
Drug interactions are minimal with fluoroquinolones, which do not affect the cytochrome P450 system, with the exception of the CYP1A2 enzyme (responsible for metabolism of methylxanthines, caffeine, methadone, clozapine). Co-adminstration of warfarin with a fluoroquinolone can result in prolongation of the prothrombin time. Absorption of the oral fluoroquinolones is also decreased by co-administration of divalent cations, such as aluminum, calcium, iron and magnesium. Therefore, antacids and dietary supplements (as well as milk and milk-containing products) should be discontinued or avoided near the dosing interval of the fluoroquinolone to prevent suboptimal serum levels of the drug.
Nausea and diarrhea are the most commonly reported adverse effects of the fluoroquinolones. Rash is infrequent (0.4–2.8%), as is anaphylaxis. Liver enzyme elevations occur in 2–3% and are mild and reversible upon discontinuation of the drug. More serious toxicities include tendinopathy and QT prolongation. Fluoroquinolone-related tendinopathy occurs most often in the Achilles tendon and can occur anywhere from 2 hours to 6 months after the first dose. Rupture of the tendon occurs in up to 50% of cases. Risk factors for tendinopathy include kidney disease, dialysis and renal transplant. Prolongation of the QT interval is also a class effect and can lead to torsades de pointes in patients at risk. Ciprofloxacin has a lower risk of QT prolongation with consequent torsades de pointes than levofloxacin or moxifloxacin. Risk factors for fluoroquinolone-associated torsades include co-administration with another QT-prolonging drug (particularly class III or class IA antiarrhythmics), underlying cardiac disease, renal impairment, hypokalemia or hypomagnesemia, and female sex.
The utility of the fluoroquinolones for the treatment of gastrointestinal infections has been hindered by growing widespread resistance to these agents, particularly among Escherichia coli and Camplylobacter jejuni. The decision to use a fluoroquinolone to treat a gastrointestinal infection should be guided by knowledge of local resistance patterns in the area of acquisition (i.e., in cases of traveler’s diarrhea) and by the results of antimicrobial susceptibility testing when available.
Glycylcyclines
Tigecycline is a bacteriostatic agent related to the tetracycline class. It binds to the 30S ribosomal subunit and blocks entry of transfer RNA into the ribosome. The peptide chain cannot elongate, and protein synthesis is inhibited. It is available only intravenously. The volume of distribution is large, indicating extensive distribution into tissues, with highest levels in bone and bone marrow. Most of the drug is excreted unchanged by the biliary system, with smaller portions undergoing glucuronidation by the liver, and <30% excreted unchanged in the urine. No dosage adjustments are required in renal insufficiency. No dosage adjustments are recommended in mild hepatic impairment (Child A or B cirrhosis), but in Child C cirrhosis, the maintenance dose is reduced to 25 mg IV every 12 hours. Caution should be used in this population. There are no major drug interactions with tigecycline.
The most common adverse effects of tigecycline are nausea and vomiting, which occur in up to 30% of patients. These symptoms can be dose-limiting and can be lessened by administering anti-emetics or food at the time of infusion. Symptoms usually begin in the first 1–2 days of treatment and are more common in younger patients and women.
Tigecycline has broad spectrum coverage of gram-positive, gram-negative and anaerobic organisms. It retains activity against methicillin-resistant S aureus and vancomycin-resistant enterococci. It has no Pseudomonas activity, but is active against some strains of resistant Acinetobacter.
Glycopeptides
Vancomycin is a glycopeptide that works through inhibition of cell wall synthesis in dividing bacteria. Although it is most commonly used in its intravenous form for serious gram-positive infections such as methicillin-resistant S. aureus, its most common use in gastrointestinal infections is in its oral form for C. difficile. Intravenous vancomycin concentrates poorly in the stool and is ineffective in treating C. difficile.
When given orally or per rectum, vancomycin is poorly absorbed from the gastrointestinal tract, even when pseudomembranous colitis is present. It is excreted unchanged in the feces. Because of the lack of systemic absorption, drug interactions are not significant with oral administration. Adverse effects are also minimal, and oral vancomycin is well-tolerated. There is a risk of selection for vancomycin-resistant enterococci with use of oral vancomycin, but the incidence and clinical significance of this risk has not been firmly established.
Macrolides
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


