CHAPTER 62 Anatomy, Histology, Embryology, Developmental Anomalies, and Pediatric Disorders of the Biliary Tract
EMBRYOLOGY OF THE LIVER AND BILIARY TRACT
The human liver is formed from two primordia (Fig. 62-1): the liver diverticulum and the septum transversum.1 Proximity of cardiac mesoderm, which expresses fibroblast growth factors (FGFs) 1, 2, and 8, and bone morphogenetic proteins cause the foregut endoderm to develop into the liver.2 Surrounding mesoderm and ectoderm participate in the hepatic specification of the endoderm, and many transcription factors, such as cJun, retinoblastoma gene, and nuclear factor κB, play important roles as regulators of liver embryogenesis.3 The liver diverticulum forms through proliferation of endodermal cells at the cranioventral junction of the yolk sac with the foregut and grows into the septum transversum in a cranioventral direction.4 The earliest marker of mammalian hepatic differentiation is the endodermal expression of albumin, transthyretin, and alpha fetoprotein. Cells that express these markers are called hepatoblasts, and they differentiate into hepatocytes and epithelial cells of the bile ducts. Signaling mediated by the stress-activated protein kinase (SAPK)/Jun N-terminal kinase (JNK) pathway promotes hepatoblast proliferation as well as survival.5 This early change occurs on the eighteenth day of gestation and corresponds to the 2.5-mm stage of the embryo. The signaling molecules that elicit embryonic induction of the liver from the mammalian gut endoderm or induction of other gut-derived organs are being defined. The homeobox gene Hhex is essential for proper hepatoblast differentiation and bile duct morphogenesis. Members of the GATA, FOXA, ONECUT1, and hepatocyte nuclear factor (HNF)3/forkhead transcription factor families are also required for the formation and differentiation of gut endoderm tissues.3,4 The septum transversum consists of mesenchymal cells and a capillary plexus formed by the branches of the two vitelline veins. At the 3- to 4-mm stage, between the third and fourth weeks of gestation, the growing diverticulum projects as an epithelial plug into the septum transversum.5 The homeodomain transcription factors Hex and Prox1, expressed in the anterior endoderm and hepatic diverticulum, are required for migration of hepatoblasts into the septum transversum that precedes liver growth and morphogenesis.6,7 Another homeodomain protein, Hlx, is necessary for hepatoblast proliferation. At the 5-mm stage, a solid cranial portion (hepatic) and a hollow caudal portion of the diverticulum can be clearly distinguished. The large hepatic portion differentiates into proliferating cords of hepatocytes and the intrahepatic bile ducts. HNF4a expression drives further hepatocyte differentiation and epithelial transformation into the characteristic sinusoidal architecture.8 The smaller, cystic portion, which initially is a cord of epithelial cells, forms the gallbladder, bile duct, and cystic duct through a process of elongation and recanalization.
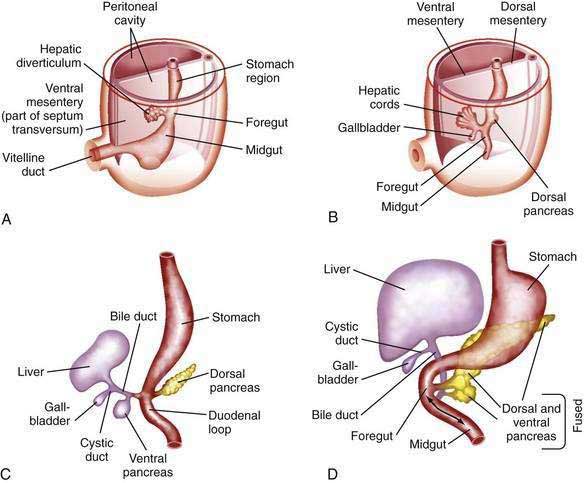
The intrahepatic bile ducts develop from primitive hepatocytes around branches of the portal vein. Cholangiocytes are associated with the basement membrane throughout bile duct development, suggesting that cholangiocytes receive morphogenic signals from components of the extracellular matrix including laminin and type IV collagen.9,10 A ring of hepatocytes in proximity to the portal vein branches first transforms into bile duct–type cells. A second layer of primitive hepatocytes is similarly transformed and produces a circular cleft around the portal vein that is lined on both sides by bile duct epithelial cells.11 This double-walled cylinder with a slit-like lumen, the ductal plate, can be detected at 9 weeks of gestation. Thus, the entire network of interlobular and intralobular bile ductules develops from the limiting plate. The transcription factors Hes1, HNF6, and HNF1β are required for gallbladder and bile duct development.6 The Notch and transforming growth factor-β (TGF-β) signaling pathways are activated in hepatoblasts surrounding the portal veins, allowing hepatoblasts to become cholangiocytes.5 In sections of the 10-mm embryo, many of the liver cords are traversed by double-walled canals that branch and morphologically are indistinguishable from bile capillaries of the adult. These structures differ from those of the adult in that they are bounded by six or more liver cells instead of two. The process of differentiation of bile ductular epithelial cells (cholangiocytes) from primitive hepatocytes has been documented in humans through the use of immunohistochemical staining with several anticytokeratin antibodies. During the phenotypic shift toward bile duct–type cells, hepatocytes first display increased reactivity for cytokeratins 8 and 18 and express cytokeratin 19 at 20 to 25 weeks of gestation.12 Cholangiocyte-mesenchymal cell interaction is important for the formation of bile ducts. During the transition from ductal plates to bile ducts, portal myofibroblasts significantly expand and surround newly formed bile ducts. Periportal connective tissue, corticosteroid hormones, and basal laminar components may play important roles in the differentiation of bile ducts. The ductal plate structure requires extensive remodeling through a process of reabsorption, possibly through apoptosis, to yield the characteristic anastomosing system of biliary channels that surround the portal vein. Proteins that appear to have a role in the promotion of apoptosis, specifically Fas antigen and c-myc, are consistently detected in primitive intrahepatic ductal cells.5 Lewis antigen, which is expressed in damaged and apoptotic cells, is also present. Bcl-2 protein, an inhibitor of apoptosis, is not found in early stages of intrahepatic bile duct cell development but becomes detectable later. Computed three-dimensional reconstruction of the developing ductal plate has shown that the ductal plate remodeling process starts at the porta hepatis at approximately 11 weeks of gestation and progresses toward the periphery of the liver.12 The process is in large part completed at term, but even at 40 weeks of gestation, some of the smallest portal vein branches may not be accompanied by an individual bile duct and may still be surrounded by a (discontinuous) ductal plate. In ductal plate malformation, which occurs in biliary disorders such as congenital hepatic fibrosis and Caroli’s disease (see later), insufficient reabsorption of ductal plates can result in the formation of large dilated segments of a primitive bile duct that surrounds the central portal vein.12
The gallbladder and extrahepatic bile ducts start to develop from hepatic endodermal cells and hepatoblasts immediately after formation of the liver primordium. Foxf1 is critical for mesenchymal epithelial cell induction of gallbladder morphogenesis.6 In embryos 5 to 6 mm in length, the original hepatic diverticulum differentiates cranially into proliferating hepatic cords and bile ducts and caudally into the gallbladder. The cystic portion of the liver diverticulum is hollow initially, but the lumen is filled as cells continually migrate into it. A study in 1994 showed that the primitive extrahepatic bile duct maintains continuity with the ductal plate, from which intrahepatic bile ducts are eventually formed.9,10 Contrary to long-held concepts of biliary development, no “solid stage” of endodermal occlusion of the bile duct lumen is found at any stage of gestation. At 16 mm, the cystic duct and proximal gallbladder are hollow, but the fundus of the gallbladder is still partially obstructed by remnants of the epithelial plug. The gallbladder is patent by the third month of gestation. Further development, until birth, consists primarily of continued growth. The characteristic folds of the gallbladder are formed toward the end of gestation and are moderately developed in the neonate. Bile secretion starts at the beginning of the fourth month of gestation; thereafter, the biliary system continuously contains bile, which is secreted into the gut and imparts a dark green color to the intestinal contents (meconium).
ANATOMY
BILE DUCTS
The adult human liver has more than 2 km of bile ductules and ducts. Quantitative computer-aided three-dimensional imaging has estimated the volume of the entire macroscopic duct system of human liver to be a mean of 20.4 cm.3,13 In these studies the mean internal surface of 398 cm2 is magnified approximately 5.5-fold by the presence of microvilli and cilia at the apical surface of cholangiocytes that play an important role in the regulation of cholangiocyte functions. These structures are far from being inert channels; they are capable of modifying biliary flow and composition significantly in response to hormones such as secretin.14,15 A general feature of bile ductules is their anatomic intimacy with portal blood and lymph vessels, which potentially allows selective exchange of materials between compartments. No major ultrastructural differences exist between cholangiocytes lining small and large bile ducts, but the functional properties of cholangiocytes are heterogeneous.15 For example, large, but not small, intrahepatic bile ducts are involved in secretin-regulated bile ductal secretion.16 Correspondingly, the secretin receptor and chloride-bicarbonate exchanger messenger ribonucleic acids (mRNAs) have been detected in large, but not small, intrahepatic bile duct units.15
Bile secretion begins at the level of the bile canaliculus, the smallest branch of the biliary tree.17 Its boundaries are formed by a specialized membrane of adjacent apical poles of liver cells. The canaliculi form a meshwork of polygonal channels between hepatocytes with many anastomotic interconnections.17 Bile then enters the small terminal channels (the canals of Hering), which have a basement membrane and are lined partly by hepatocytes and partly by cholangiocytes.13 The canals of Hering provide a conduit through which bile may traverse the limiting plate of hepatocytes to enter the larger perilobular or intralobular ducts.18,19 These smallest of biliary radicles are less than 15 to 20 µm in diameter with lumens surrounded by cuboidal epithelial cells. At the most proximal level, one or more fusiform-shaped ductular cells may share a canalicular lumen with a hepatocyte; gradually, the ductules become lined by two to four cuboidal epithelial cells as they approach the portal canal.17 Bile flows from the central lobular cells toward portal triads (from zone 3 to zone 1 of the liver acinus) (see Chapter 71). The terminal bile ductules are thought to proliferate as a result of chronic extrahepatic bile duct obstruction.19
The interlobular bile ducts form a richly anastomosing network that closely surrounds the branches of the portal vein.20–22 These bile ducts (Fig. 62-2) are initially 30 to 40 µm in diameter and are lined by a layer of cuboidal or columnar epithelium that displays a microvillar architecture on its luminal surface.17 The cells have a prominent Golgi apparatus and numerous vesicles that likely participate in the exchange of substances among cytoplasm, bile, and plasma through the processes of exocytosis and endocytosis.17 These ducts increase in caliber and possess smooth muscle fibers within their walls as they approach the hilum of the liver. The muscular component may provide the morphologic basis for the narrowing of the ducts at this level, as observed on cholangiography.22 Furthermore, as the ducts become progressively larger, the epithelium becomes thicker, and the surrounding layer of connective tissue grows thicker and contains many elastic fibers. These ducts anastomose further to form the large hilar, intrahepatic ducts, which are 1 to 1.5 mm in diameter and give rise to the main hepatic ducts.
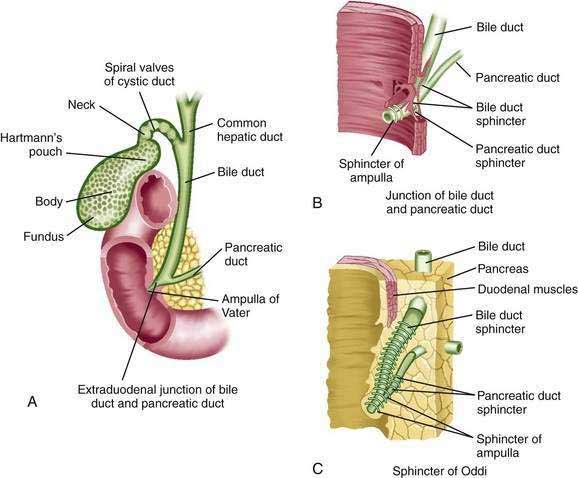
The common hepatic duct emerges from the porta hepatis after the union of the right and left hepatic ducts, each of which is 0.5 to 2.5 cm long (Fig. 62-3).23,24 The confluence of the right and left hepatic ducts is outside the liver in approximately 95% of cases; uncommonly, the ducts merge inside the liver, or the right and left hepatic ducts do not join until the cystic duct joins the right hepatic duct.24 As the hepatic ducts leave the porta hepatis, they lie within the two serous layers of the hepatoduodenal ligament. This sheath of fibrous tissue binds the hepatic ducts to the adjacent blood vessels. In the adult, the common hepatic duct is approximately 3 cm long and is joined by the cystic duct, usually at its right side, to form the bile duct (or common bile duct).24 However, the length and angle of junction of the cystic duct with the common hepatic duct are variable. The cystic duct enters the common hepatic duct directly in 70% of patients; alternatively, the cystic duct may run anterior or posterior to the bile duct and spiral around it before joining the bile duct on its medial side.23 The cystic duct may also course parallel to the common hepatic duct for 5 to 6 cm and enter it after running posterior to the first portion of the duodenum.
In humans, the large intrahepatic bile ducts at the hilum (1- to 1.5-mm diameter) have many irregular side branches and pouches (150- to 270-µm diameter) that are oriented in one plane, corresponding anatomically to the transverse fissure.17 Smaller pouches of the side branches are also found. Many side branches end as blind pouches, but others, particularly at the hilum, communicate with each other. At the bifurcation, side branches from several main bile ducts connect to form a plexus. The functional significance of these structures is not known. The blind pouches may serve to store or modify bile, whereas the biliary plexus provides anastomoses, which may allow exchange of material between the large bile ducts.
The anatomy of the hepatic hilum is particularly important to the surgeon. A plate of fibrous connective tissue in the hepatic hilum includes the umbilical plate that envelops the umbilical portion of the portal vein, the cystic plate in the gallbladder bed, and the Arantian plate that covers the ligamentum venosum.24 Histologic examination of the sagittal section of the hilar plate reveals abundant connective tissue, including neural fibers, lymphatic vessels, small capillaries, and small bile ducts. The bile ducts in the plate system correspond to the extrahepatic bile ducts, and their lengths are variable for every segment.24
Like the intestine, the cystic, common hepatic, and bile ducts possess mucosa, submucosa, and muscularis.22 The ducts are lined by a single layer of columnar epithelium. Mucus secreting tubular glands can be found at regular intervals in the submucosa, with openings to the surface of the mucosa. The bile duct is approximately 7 cm long, runs between layers of the lesser omentum, and lies anterior to the portal vein and to the right of the hepatic artery.24 The bile duct normally is 0.5 to 1.5 cm in diameter.19 The wall of the extrahepatic bile duct is supported by a layer of connective tissue with an admixture of occasional smooth muscle fibers. The smooth muscle component is conspicuous only at the neck of the gallbladder and at the lower end of the bile duct. The bile duct passes retroperitoneally behind the first portion of the duodenum in a notch on the back of the head of the pancreas and enters the second part of the duodenum. The duct then passes obliquely through the posterior medial aspect of the duodenal wall and joins the main pancreatic duct to form the ampulla of Vater (see Fig. 62-3).23 The mucous membrane bulge produced by the ampulla forms an eminence, the duodenal papilla. In approximately 10% to 15% of patients, the bile and pancreatic ducts open separately into the duodenum. The bile duct tapers to a diameter of 0.6 cm or less before its union with the pancreatic duct.24
As they course through the duodenal wall, the bile and pancreatic ducts are invested by a thickening of both the longitudinal and circular layers of smooth muscle (see Fig. 62-3) of the sphincter of Oddi.25 There is considerable variation in this structure, but it is usually composed of several parts: (1) the sphincter choledochus—circular muscle fibers that surround the intramural portion of the bile duct immediately before its junction with the pancreatic duct; (2) the sphincter pancreaticus, which is present in approximately one third of individuals and surrounds the intraduodenal portion of the pancreatic duct before its juncture with the ampulla; (3) the fasciculi longitudinales—longitudinal muscle bundles that span intervals between the bile and pancreatic ducts; and (4) the sphincter ampullae—longitudinal muscle fibers that surround a sparse layer of circular fibers around the ampulla of Vater.22 The sphincter choledochus constricts the lumen of the bile duct and thus prevents the flow of bile. Contraction of the fasciculi longitudinales shortens the length of the bile duct and thus promotes the flow of bile into the duodenum. The contraction of the sphincter ampullae shortens the ampulla and approximates the ampullary folds to prevent reflux of intestinal contents into the bile and pancreatic ducts. When both ducts end in the ampulla, however, contraction of the sphincter may cause reflux of bile into the pancreatic duct.25
The arterial supply of the bile ducts arises mainly from the right hepatic artery.20 An extraordinarily rich plexus of capillaries surrounds bile ducts as they pass through the portal tracts.20,26 Blood flowing through this peribiliary plexus empties into the hepatic sinusoids via the interlobular branches of the portal vein. The peribiliary plexus may modify biliary secretions through the bidirectional exchange of proteins, inorganic ions, and bile acids between blood and bile. Because blood flows in the direction (from the large toward the small ducts) opposite to that of bile flow, the peribiliary plexus presents a countercurrent stream of biliary-reabsorbed substances to hepatocytes.
An abundant anastomotic network of blood vessels from branches of the hepatic and gastroduodenal arteries supplies the bile duct.22,26 The supraduodenal portion of the duct is supplied by vessels running along its wall inferiorly from the retroduodenal artery and superiorly from the right hepatic artery. Injury to these blood vessels can result in bile duct stricturing.23
The lymphatic vessels of the hepatic, cystic, and proximal portions of the bile duct empty into glands at the hilum of the liver.22 Lymphatics draining from the lower portion of the bile duct drain into glands near the head of the pancreas.
GALLBLADDER
The gallbladder (see Fig. 62-3) is a storage reservoir that allows bile acids to be delivered in a high concentration and in a controlled manner to the duodenum for the solubilization of dietary lipid (see Chapter 64).22,27 It lies in a fossa on the undersurface of the right lobe of the liver.27 This distensible pear-shaped structure is 3 cm wide and 7 cm long in the adult and has a capacity of 30 to 50 mL.27 The gallbladder has a thin muscular layer with the smooth muscle cells largely oriented around the circumference of the gallbladder. The absorptive surface of the gallbladder is enhanced by numerous prominent folds. The gallbladder is covered anteriorly by an adventitia that is fused with the capsule of the liver. On its posterior aspect and at the apex, it is covered by the visceral peritoneum. The portions of the gallbladder are the fundus, body, infundibulum, and neck.22 The anterior portion of the fundus is located at the level of the right lateral border of the musculus rectus abdominis and the ninth costal cartilage. The posterior aspects of the fundus and body lie close to the transverse colon and duodenum, respectively. Thus, with perforation of the gallbladder, gallstones can readily penetrate these structures.27,28 The infundibulum is an area of tapering between the gallbladder body and neck. Hartmann’s pouch is a bulging of the inferior surface of the infundibulum that lies close to the neck of the gallbladder. Gallstones can become impacted in Hartmann’s pouch, thereby obstructing the cystic duct and producing cholecystitis.27 Extensive inflammation in Hartmann’s pouch can lead to obstruction of the adjacent common hepatic duct (Mirizzi’s syndrome).
The gallbladder is connected at its neck to the cystic duct, which empties into the bile duct (see Fig. 62-3).27 The cystic duct is approximately 4 cm long and maintains continuity with the surface columnar epithelium, lamina propria, muscularis, and serosa of the gallbladder. The mucous membrane of the gallbladder neck forms the spiral valve of Heister, which is involved in regulating flow into and out of the gallbladder.
The gallbladder is supplied by the cystic artery, which usually arises from the right hepatic artery.27,29 The artery divides into two branches near the neck of the gallbladder: a superficial branch that supplies the serosal surface and a deep branch that supplies the interior layers of the gallbladder wall. Variations in the origin and course of the cystic artery are common.27 Because the cystic artery is an end artery, the gallbladder is particularly susceptible to ischemic injury and necrosis that result from inflammation or interruption of hepatic arterial flow.
The cystic vein provides venous drainage from the gallbladder and cystic ducts and commonly empties into the portal vein and occasionally directly into the hepatic sinusoids.22,27 The lymph vessels of the gallbladder are connected with the lymph vessels of Glisson’s capsule. Subserosal and submucosal lymphatics empty into a lymph gland near the neck of the gallbladder.22 The sympathetic innervation of the gallbladder originates from the celiac axis and travels with branches of the hepatic artery and portal vein. Visceral pain is conducted through sympathetic fibers and is frequently referred to the right subcostal, epigastric, and right scapular regions. Branches of both vagus nerves provide parasympathetic innervation that likely contributes to the regulation of gallbladder motility.22
The gallbladder is lined by a mucosa that manifests multiple ridges and folds and is composed of a layer of columnar epithelial cells. The gallbladder wall consists of a mucosa, lamina propria, tunica muscularis, and serosa.30 The tunica muscularis is thick and invested with an interlocking array of longitudinal and spiral smooth muscle fibers. Tubuloalveolar glands are found in the region of the neck of the gallbladder and are involved in the production of mucus.27,30 The Rokitansky-Aschoff sinuses are invaginations of the surface epithelium that may extend through the muscularis.22 These structures can be a source of inflammation, most likely as a result of bacterial stasis and proliferation within the invaginations. The ducts of Luschka may be observed along the hepatic surface of the gallbladder and open directly into the intrahepatic bile ducts rather than into the gallbladder cavity. These structures are thought to represent a developmental anomaly, and when they are present in the gallbladder bed may be a source of a bile leak after cholecystectomy.27
CONGENITAL ANOMALIES OF THE EXTRAHEPATIC DUCTS
Accessory bile ducts are aberrant ducts that drain individual segments of the liver; they may drain directly into the gallbladder, cystic duct, right and left hepatic ducts, or bile duct.23,31 In rare cases, the right hepatic duct may connect to the gallbladder or cystic duct. These anomalies must be recognized on cholangiography in order to prevent inadvertent transection or ligation of bile ducts during surgery.
Complete duplication of the bile duct occurs rarely. In most cases, separate ducts drain the right and left hepatic lobes and open into the duodenum.23
Variation in the drainage and course of the cystic duct is common.23 Duplication of the cystic duct may also be encountered. The cystic duct is absent in most cases of agenesis of the gallbladder; rarely the duct alone may be absent, and the gallbladder empties directly into the common hepatic duct.
CONGENITAL ANOMALIES OF THE GALLBLADDER
A number of structural anomalies of the gallbladder have been described.23,31 Most of these defects are of no clinical importance, but occasionally the abnormal gallbladder may be a predisposing factor for bile stasis, inflammation, and formation of gallstones. Gallbladder disease in an anomalous or a malpositioned gallbladder may cause diagnostic confusion.
Agenesis of the gallbladder may be an isolated anomaly or occur in association with other congenital malformations.31 The abnormality has a frequency at autopsy of 0.04% to 0.13% and likely reflects a lack of development of the gallbladder bud or failure of the normal process of vacuolization. Incomplete vacuolization of the solid endodermal cord during development can result in congenital strictures of the gallbladder or cystic duct. Biliary atresia is commonly associated with an absent or atretic gallbladder. Hypoplasia of the gallbladder has been described, particularly in patients with cystic fibrosis. Ectopic tissues of foregut endodermal origin, including gastric, hepatic, adrenal, pancreatic, and thyroid tissues, may be found within the gallbladder wall.
A double gallbladder is another rare malformation, which occurs in approximately 1 to 5 per 10,000 persons in the general population.31,32 The two gallbladders may share a single cystic duct, forming a Y-shaped channel, or each may have a distinct cystic duct that enters the bile duct separately.23 Vesica fellae triplex, or triplication of the gallbladder, is another rare congenital anomaly.33 Multiple gallbladders are usually discovered because of cholelithiasis, sludge, cholecystitis, or neoplasia. Bilobed gallbladders and gallbladder diverticula are other rare anomalies. A single gallbladder may be divided by longitudinal septa into multiple chambers, probably secondary to incomplete vacuolization of the solid gallbladder bud during morphogenesis.32 Diverticula and septations of the gallbladder may promote bile stasis and gallstone formation.
Various malpositions of the gallbladder have been described.32 Rarely, the gallbladder lies under the left lobe of the liver, to the left of the falciform ligament. This defect likely results from migration of the embryonic bud from the hepatic diverticula to the left rather than to the right.23 Some researchers have proposed that the second gallbladder may develop independently from the left hepatic duct, with regression of the normal structure on the right. In other cases, a caudal bud that advances farther than the cranial bud may become buried within the cranial structure, creating an intrahepatic gallbladder. It is thought that if the caudal bud lags behind the movement of the cranial bud, a floating gallbladder results. In this setting, the gallbladder is covered completely with peritoneum and suspended from the undersurface of the liver by mesentery to the gallbladder or cystic duct; the gallbladder is abnormally mobile and prone to torsion. Rarely, gallbladders have been found in the abdominal wall, falciform ligament, and retroperitoneum.32
Several forms of “folded” gallbladders have been described. In one variant, the fundus appears to be bent, giving the appearance of a “Phrygian cap.”32 The gallbladder is usually located in a retroserosal position, and the anomaly is thought to result from aberrant folding of the gallbladder within the embryonic fossa. Aberrant folding of the fossa during the early stages of development can result in kinking between the body and the infundibulum of the gallbladder. Kinked gallbladders probably do not lead to clinical symptoms but may be a source of confusion in the interpretation of imaging studies.32
AN OVERVIEW OF DISORDERS OF THE BILIARY TRACT IN INFANTS AND CHILDREN
Cholestatic liver disease results from processes that interfere with either bile formation by hepatocytes or bile flow through the intrahepatic and extrahepatic biliary tree. A number of these disorders result from defective ontogenesis as well as from a failure of postnatal adaptation to the extrauterine environment. Table 62-1 provides a list of disorders that affect the biliary tract and occur in both infants and older children and that are discussed later in the chapter. There is a particular emphasis on neonatal cholangiopathies and the unique aspects of biliary disease in the older child. The general features of the many cholestatic liver diseases of the neonate are similar, and a central problem of pediatric hepatology is differentiating intrahepatic from extrahepatic cholestasis (Table 62-2).34 The treatment of metabolic or infective liver diseases and the surgical management of biliary anomalies require early diagnosis. Even when effective treatment is not possible, infants and children with progressive liver disease benefit from optimal nutritional support and medical management of chronic liver disease before they are referred for liver transplantation.
Table 62-1 Disorders of the Biliary Tract in Infants and Children
Modified from Balistreri WF. Neonatal cholestasis: Lessons from the past, issues for the future. Semin Liver Dis 1987; 7:61-6.
Table 62-2 Relative Frequencies of Various Forms of Neonatal Cholestasis
| DISORDER | FREQUENCY |
|---|---|
| Idiopathic neonatal hepatitis | 30-35 |
| Extrahepatic biliary atresia | 30 |
| α1-Antitrypsin deficiency | 7-10 |
| Intrahepatic cholestatic syndromes (Alagille syndrome, Byler’s disease, others) | 5-6 |
| Hepatitis (cytomegalovirus, rubella, herpes simplex virus, others) | 3-5 |
| Choledochal cyst | 2-4 |
| Bacterial sepsis | 2 |
| Endocrinopathy (hypothyroidism, panhypopituitarism) | ≈1 |
| Galactosemia | ≈1 |
| Inborn errors of bile acid metabolism | ≈1 |
| Other metabolic disorders | ≈1 |
Modified from Balistreri WF. Neonatal cholestasis: Lessons from the past, issues for the future. Semin Liver Dis 1987; 7:61.
Because of the immaturity of hepatobiliary function, the number of distinct disorders that exhibit cholestatic jaundice may be greater during the neonatal period than at any other time of life (see Table 62-1).35,36 Liver dysfunction in the infant, regardless of the cause, is commonly associated with bile secretory failure and cholestatic jaundice. Although cholestasis may be traced to the level of the hepatocyte or the biliary apparatus, in practice there may be considerable overlap among disorders with regard to the initial and subsequent sites of injury. For example, damage to the biliary epithelium often is a prominent feature of neonatal hepatitis that results from cytomegalovirus infection. Mechanical obstruction of the biliary tract invariably produces liver dysfunction and in the neonate may be associated with abnormalities of the liver parenchyma, such as giant cell transformation of hepatocytes. Whether giant cells, a frequent, nonspecific manifestation of neonatal liver injury, reflect the noxious effects of biliary obstruction or whether the hepatocytes and the biliary epithelium are damaged by a common agent during ontogenesis, such as a virus with tropism for both types of cells, is unknown. Furthermore, another common histologic variable that often accompanies neonatal cholestasis is bile ductular paucity or a diminution in the number of interlobular bile ducts.37 This finding may be of primary importance in patients with syndromic paucity of intrahepatic bile ducts but may also occur as an occasional feature of many other disorders, including idiopathic neonatal hepatitis, congenital cytomegalovirus infection, and α1-antitrypsin deficiency.38 Serial liver biopsies usually show a progressive decrease in the number of bile ductules per portal tract, with a variable amount of associated inflammation.
DIAGNOSIS
In most infants with cholestatic liver disease the condition appears during the first few weeks of life. Differentiating conjugated hyperbilirubinemia from the common unconjugated, physiologic hyperbilirubinemia of the neonate or the prolonged jaundice occasionally associated with breast-feeding is essential.39 The possibility of liver or biliary tract disease must be considered in any neonate older than 14 days with jaundice. The stools of a patient with well-established biliary atresia are acholic; however, early in the course of incomplete or evolving biliary obstruction, the stools may appear normal or only intermittently pigmented. Life-threatening but treatable disorders such as bacterial infection and a number of inborn errors of metabolism must be excluded. Furthermore, the success of surgical procedures in relieving the biliary obstruction of biliary atresia or a choledochal cyst depends on early diagnosis and surgery.
The approach to the evaluation of an infant with cholestatic liver disease is outlined in Table 62-3. The initial assessment should establish promptly whether cholestatic jaundice is present and assess the severity of liver dysfunction. A more detailed investigation may be required and should be guided by the clinical features of the case. All relevant diagnostic tests need not be performed in every patient. For example, ultrasonography may promptly establish a diagnosis of a choledochal cyst in a neonate with jaundice and thus obviate the need to exclude infectious and metabolic causes of liver disease. Numerous routine and specialized biochemical tests and imaging procedures have been proposed to distinguish intrahepatic from extrahepatic cholestasis in infants and thereby avoid unnecessary surgical exploration.39,40 Standard liver biochemical tests usually show variable elevations in serum direct bilirubin, aminotransferase, alkaline phosphatase, and lipid levels. Unfortunately, no single test has proved to have satisfactory discriminatory value, because at least 10% of infants with intrahepatic cholestasis have bile secretory failure sufficient to lead to an overlap in diagnostic test results with those suggestive of biliary atresia.41 The presence of bile pigment in stools is sometimes cited as evidence against biliary atresia, but coloration of feces with secretions and epithelial cells that have been shed by the cholestatic patient may be misleading.
Table 62-3 Evaluation of the Infant with Cholestasis
Tests to Establish the Presence and Severity of Liver Disease
ALT, alanine aminotransferase; AST, aspartate aminotransferase; EBV, Epstein-Barr virus; HBsAg, hepatitis B surface antigen; MRCP, magnetic resonance cholangiopancreatography; STS, serologic test for syphilis; TORCH, toxoplasmosis, rubella, cytomegalovirus, herpesvirus.
Ultrasonography can be used to assess the size and echogenicity of the liver. Even in neonates, high-frequency, real-time ultrasonography usually can define the presence and size of the gallbladder, detect stones and sludge in the bile ducts and gallbladder, and demonstrate cystic or obstructive dilatation of the biliary system.42,43 Extrahepatic anomalies also may be identified. A triangular cord or bandlike periportal echogenicity (3 mm or greater in thickness), which represents a cone-shaped fibrotic mass cranial to the portal vein, appears to be a specific ultrasonographic finding in the early diagnosis of biliary atresia.42,43 The gallbladder “ghost” triad, defined as gallbladder length less than 1.9 cm, lack of smooth or complete echogenic mucosal lining with an indistinct wall, and irregular or lobular contour, has been proposed as additional criteria for biliary atresia.
Computed tomography provides information similar to that obtained by ultrasonography but is less suitable in patients younger than 2 years because of exposure to radiation, the paucity of intra-abdominal fat for contrast, and the need for heavy sedation or general anesthesia.44
Magnetic resonance cholangiopancreatography (MRCP), performed with T2-weighted turbo-spin echo sequences, is widely used to assess the biliary tract in all age groups. In a 1999 study, MRCP reliably demonstrated the bile duct and gallbladder in normal neonates. In some patients with biliary atresia, nonvisualization of the bile duct and demonstration of a small gallbladder have been characteristic MRCP findings.45 A more recent study found that MRCP is 82% accurate, 90% sensitive, and 77% specific for depicting extrahepatic biliary atresia. Contrary to previous reports, false-positive and false-negative findings occur with MRCP. Differentiation of severe intrahepatic cholestasis from biliary atresia may be difficult because the ability of MRCP to delineate the extrahepatic biliary tree depends on bile flow.46
The use of hepatobiliary scintigraphic imaging agents such as 99mTc iminodiacetic acid derivatives may be helpful in differentiating extrahepatic biliary atresia from other causes of neonatal jaundice.44 Unfortunately, a 1997 study showed that in 50% of patients who had a paucity of interlobular bile ducts but no extrahepatic obstruction, biliary excretion of radionuclide was absent.47 Twenty-five percent of patients who had idiopathic neonatal hepatitis also demonstrated no biliary excretion. Nevertheless, the modality remains useful for assessing cystic duct patency in patients with a hydropic gallbladder or cholelithiasis.
Percutaneous transhepatic cholangiopancreatography may be of value in visualizing the biliary tract in selected patients,48 but the technique is more difficult to perform in infants than in adults because the intrahepatic bile ducts are small and because most disorders that occur in infants do not result in dilatation of the biliary tree. Endoscopic retrograde cholangiopancreatography (ERCP) may be useful in evaluating children with extrahepatic biliary obstruction and has been performed successfully in a small number of cholestatic neonates.49 Considerable technical expertise is required of the operator to complete this procedure in infants. Most neonates require general anesthesia for a satisfactory examination.
Percutaneous liver biopsy is particularly valuable in evaluating cholestatic patients and can be undertaken in even the smallest infants with only sedation and local anesthesia.50 For example, a diagnosis of extrahepatic biliary atresia can be made on the basis of clinical and histologic criteria in 90% to 95% of patients. When doubt about the diagnosis persists, the patency of the biliary tree can be examined directly by a minilaparotomy and operative cholangiogram.
PEDIATRIC DISORDERS OF THE BILE DUCTS
BILIARY ATRESIA
Biliary atresia is characterized by the complete obstruction of bile flow as a result of the destruction or absence of all or a portion of the extrahepatic bile ducts.51 As part of the underlying disease process or as a result of biliary obstruction, concomitant injury and fibrosis of the intrahepatic bile ducts also occurs to a variable extent. The disorder occurs in 1 in 10,000 to 15,000 live births and accounts for approximately one third of cases of neonatal cholestatic jaundice (see Table 62-2). It is the most frequent cause of death from liver disease and reason for referral for liver transplantation in children (approximately 50% of all cases).52 The cause of biliary atresia is unknown. The disease is not inherited, and there have been several reports of dizygotic and monozygotic twins discordant for biliary atresia.53 In a study of 461 patients in France, seasonality, time clustering, and time-space clustering could not be demonstrated.54 Reports of familial cases have been rare; in most, a detailed histologic description of the extrahepatic biliary tree was not provided to exclude narrowing or hypoplasia of the bile duct associated with severe intrahepatic cholestasis. In the multistate case-controlled National Birth Defects Prevention Study conducted between 1997 and 2002, babies born to non-Hispanic black mothers were at greater risk than non-Hispanic white mothers. Conception during the spring and low dietary intakes of vitamin E, copper, phosphorus, and beta tocopherol were additional risk factors.55
Several mechanisms have been proposed to account for the progressive obliteration of the extrahepatic biliary tree.56 There is no evidence that biliary atresia results from a failure in morphogenesis or recanalization of the bile duct during embryonic development. Clinical features support the concept that in most cases, injury to the biliary tract occurs after birth. There is little support for an ischemic or toxic origin of extrahepatic bile duct injury.
Congenital infections with cytomegalovirus, rubella virus, human herpesvirus 6, and papillomavirus occasionally have been implicated.56 Reovirus type 3 has been implicated on the basis of the serologic evaluation of patients and immunolocalization of reovirus 3 antigens in a bile duct remnant of a patient with biliary atresia.57,58 The results of studies on the role of reovirus in biliary atresia have been contradictory. In a 1998 report, reovirus RNA was detected by reverse-transcriptase polymerase chain reaction methodology in hepatic or biliary tissues, or both, in 55% of patients who had biliary atresia and 78% of patients who had a choledochal cyst,59 compared with 21% of patients who had other hepatobiliary diseases and 12% of autopsy controls. Initial reports of the involvement of group C rotavirus in biliary atresia have not been confirmed.60
A significant increase in human leukocyte antigen (HLA) B12 has been found among patients with biliary atresia who had no associated anomalies.61 The HLA haplotypes A9, B5, A28, and B35 have been found more frequently. Oligonucleotide-based gene chip analysis of cRNA from livers of infants with biliary atresia has demonstrated a coordinated activation of genes involved in lymphocyte differentiation and inflammation.62 The finding of overexpression of osteopontin and γ-interferon indicates a potential role of type 1 T helper (Th1)–like cytokines in the pathogenesis. Biliary atresia is associated with oligoclonal expansions of CD4+ and CD8+ T cells within liver and extrahepatic bile duct remnant tissues, indicating the presence of activated T cells that react to specific antigenic stimulation.63 In a Rhesus rotavirus (RRV) murine model of biliary atresia, γ-interferon was particularly important in mediating bile duct injury.64 In other studies adoptive transfer of T cells from RRV-diseased mice into naïve syngeneic severe combined immunodeficient (SCID) recipients, at a time when viral infection could no longer be demonstrated, caused bile duct specific inflammation, possibly in response to bile duct autoantigens.65 Circulating markers of inflammation persist in biliary atresia and are largely unaffected by portoenterostomy (see later), with clear progressive elevation in both Th1 effectors interleukin (IL)-2 and interferon, some Th2 effectors (IL-4), as well as the macrophage marker (tumor necrosis factor-α [TNF-α]). Increased expression of soluble cell adhesion molecules, sICAM-1 and sVCAM-1, are also found and likely reflect ongoing recruitment of circulating inflammatory or immunocompetent cells into target tissues.66 Whether this immune response is induced by a viral infection or reflects a genetically programmed response to an infectious or environmental exposure remains unknown.
Extrahepatic anomalies occur in 10% to 25% of patients and include cardiovascular defects, polysplenia, malrotation, situs inversus, and bowel atresias.67,68 Some patients who have heterotaxia, including an infant with biliary atresia and polysplenia, have been found to have loss-of-function mutations in the CFC1 gene.69,70 This gene encodes a protein called CRYPTIC, which is involved in establishing the left-right axis during morphogenesis. In contrast, limited studies of infants with biliary atresia and heterotaxia have not found mutations in the INV gene, which is also involved in determining laterality.71 In a microarray analysis of liver tissue from infants with a so-called embryonic form of biliary atresia in which extrahepatic malformations and early onset of cholestatic jaundice occur, a unique pattern of expression of genes involved in chromatin integrity and function (Smarca-1, Rybp, and Hdac3) and overexpression of five imprinted genes (Igf2, Peg3, Peg10, Meg3, and IPW) was found, implying a failure to down-regulate embryonic gene programs that influence the development of the liver and other organs.72 Jagged1 (the gene defective in Alagille syndrome [see later]) missense mutations were identified in 9 of 102 patients with biliary atresia and were associated with a poor prognosis.73
Pathology
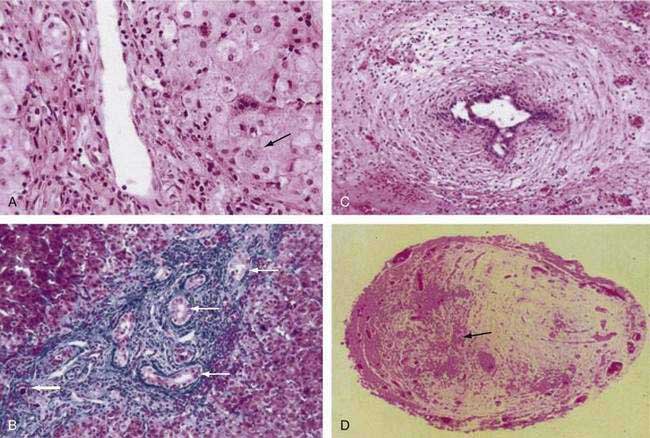
Histopathologic findings on initial liver biopsy specimens are of great importance in the management of patients with biliary atresia.51,52 Early in the course, hepatic architecture is generally preserved, with a variable degree of bile ductular proliferation, canalicular and cellular bile stasis, and portal tract edema and fibrosis (Fig. 62-4).52 The presence of bile plugs in portal triads is highly suggestive of large duct obstruction. Furthermore, bile ductules show varying injury to the biliary epithelium, including swelling, vacuolization, and even sloughing of cells into the lumen. Portal tracts may be infiltrated with inflammatory cells, and in approximately 25% of patients there may be giant cell transformation of hepatocytes to a degree observed more commonly in neonatal hepatitis. Bile ductules occasionally may assume a ductal plate configuration suggesting that the disease has interfered with the process of ductular remodeling that occurs during prenatal development.74 Biliary cirrhosis may be present initially or may evolve rapidly over the first months of life, with or without the successful restoration of bile flow.75
The morbid anatomic characteristics of the extrahepatic bile ducts in biliary atresia are highly variable. Kasai proposed a useful classification of the anatomic variants.76 Three main types have been defined on the basis of the site of the atresia. Type I is atresia of the bile duct with patent proximal ducts. Type II atresia involves the hepatic duct, with cystically dilated bile ducts at the porta hepatis. In type IIa atresia, the cystic and bile ducts are patent, whereas in type IIb atresia, these structures also are obliterated. These forms of biliary atresia have been referred to as “surgically correctable” but unfortunately account for less than 10% of all cases. Ninety percent or more of patients have type III atresia, involving obstruction of the common, hepatic, and cystic ducts, without cystically dilated hilar ducts. The entire perihilar area is in a cone of dense fibrous tissue. The gallbladder is involved to some extent in approximately 80% of patients. The type III variant has been characterized as noncorrectable, in that there are no patent hepatic or dilated hilar ducts that can be used for a biliary-enteric anastomosis.
Complete fibrous obliteration of at least a portion of the extrahepatic bile ducts is a consistent feature found on microscopic examination of the fibrous remnant.76 Other segments of the biliary tree may demonstrate lumens with varying degeneration of bile duct epithelial cells, inflammation, and fibrosis in the periductular tissues (see Fig. 62-4). In most patients, bile ducts within the liver that extend to the porta hepatis are patent during the first weeks of life but are destroyed progressively, presumably by the same process that damaged the extrahepatic ducts and by the effects of biliary obstruction. In more than 20% of patients, concentric tubular ductal structures similar to those observed in ductal plate malformations are found, indicating that the disease process interfered with the normal remodeling of the biliary tract.
Clinical Features
Most infants with biliary atresia are born at term after a normal pregnancy and have a normal birth weight.56 Female infants are affected more commonly than male infants. The perinatal course is typically unremarkable. Postnatal weight gain and development usually proceed normally. Jaundice is observed by the parents or the physician after the period of physiologic hyperbilirubinemia. Prolonged jaundice may be erroneously attributed to breastfeeding.77 The possibility of liver or biliary tract disease must be considered in any neonate older than 14 days with jaundice.78 The stools of a patient with well-established biliary atresia are acholic; however, early in the course the stools may appear normally pigmented or only intermittently pigmented.
The liver is typically enlarged with a firm edge palpable 2 to 6 cm below the right costal margin.52 The spleen is usually not enlarged early in the course but becomes enlarged as portal hypertension develops. Ascites and edema are not present initially, but coagulopathy may result from vitamin K deficiency.
Laboratory studies initially reveal evidence of cholestasis, with a serum bilirubin level of 6 to 12 mg/dL, at least 50% of which is conjugated.52 Serum aminotransferase and alkaline phosphatase levels are moderately elevated. Serum gamma glutamyl transpeptidase and 5′ nucleotidase levels are also elevated.
Treatment
When the possibility of biliary atresia has been raised by clinical, pathologic, and imaging findings, exploratory laparotomy and operative cholangiography are necessary to document the site of obstruction and to direct attempts at surgical treatment.79–81
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


