Chapter 16 Amenorrhea
DEFINITIONS
In common medical usage, amenorrhea refers to the abnormal cessation of menses.1 Physiologic amenorrhea exists before puberty, during pregnancy and lactation, and after menopause. However, these physiologic causes are not included in the standard amenorrhea classifications.
Amenorrhea can be divided into two major groups based on presentation: primary or secondary amenorrhea (Table 16-1).2,3 Although many of the causes of primary and secondary amenorrhea are similar, the most likely causes, and thus the diagnostic approach, are distinct.
Table 16-1 Definitions of Primary and Secondary Amenorrhea
Primary Amenorrhea
Primary amenorrhea is the absence of menstruation in a woman who has never menstruated. Because children do not normally menstruate before puberty, the age at which primary amenorrhea is diagnosed depends on the presence or absence of secondary sexual characteristics. In the absence of increased growth or development of secondary sexual characteristics, primary amenorrhea is diagnosed when the patient has no menses by age 13. In the presence of normal growth and development of secondary sexual characteristics, the diagnosis of primary amenorrhea is reserved for patients who have no menstruation by age 15. The incidence of primary amenorrhea in the United States is less than 0.1%. The majority of patients with primary amenorrhea will be found to have either gonadal dysgenesis (49%) or müllerian agenesis (16%).4
Secondary Amenorrhea
The incidence of secondary amenorrhea not due to pregnancy, lactation, or menopause is approximately 4%.5,6 Although the list of causes for amenorrhea is quite extensive (Table 16-2), it appears that this list will continue to grow or be modified as more sophisticated genetic testing becomes available and the genetic understanding of human disease expands. The majority of patients with amenorrhea will have premature ovarian failure, hyperprolactinemia, hypothalamic amenorrhea, or polycystic ovary syndrome (PCOS).
Table 16-2 Classification of Amenorrhea, Both Primary and Secondary3
Classification
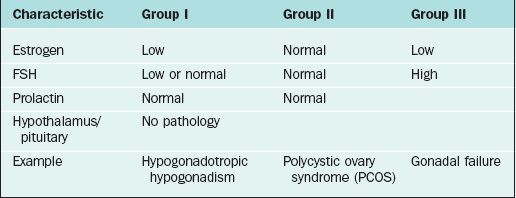
The most widely accepted classification of amenorrhea was published by the World Health Organization (WHO) and divides amenorrhea into three groups (Table 16-3).3 This WHO amenorrhea classification is designed to help the practicing clinician summarize the causes of amenorrhea to assist in evaluating the condition. Group I include individuals who lack endogenous estrogen production, in association with normal or low follicle-stimulating hormone (FSH) levels, and no evidence of hypothalamic-pituitary pathology or elevated prolactin levels. Group II is associated with evidence of estrogen production and normal levels of prolactin and FSH. Finally, Group III involves elevated serum FSH levels that indicate gonadal failure.7 Although amenorrhea can occur among patients with sexual ambiguity or virilization, it is rarely the cause for initial consultation.8
PRIMARY AMENORRHEA
The etiologies of primary amenorrhea are multiple and diverse (Tables 16-4 and 16-5). The four most common causes of primary amenorrhea have been reported to be the following:4
Table 16-4 Common Causes of Primary Amenorrhea
| Category | Frequency |
|---|---|
| Normal Secondary Sexual Development | (∼1/3 of total) |
| Müllerian agenesis | 10% |
| Androgen insensitivity | 9% |
| Constitutional delay | 8% |
| Outlet obstruction (e.g., vaginal septum, imperforate hymen) | 3% |
| Absent Secondary Sexual Development | (∼2/3 of total) |
| High FSH (gonadal dysgenesis) | |
| Abnormal karyotype (e.g., 46,XO, mosaic) | 20% |
| 46,XX | 15% |
| 46,XY | 5% |
| Low FSH | |
| Hypothalamic disorders | 8% |
| Constitutional delay | 10% |
| Hyperandrogenic conditions (e.g., PCOS, CAH) | 6% |
| Pituitary adenomas | 5% |
Adapted from Practice Committee of ASRM: Current evaluation of amenorrhea. Fertil Steril 82(Suppl 1):33-S39, 2004.
Gonadal Dysgenesis
The term gonadal dysgenesis is used globally to refer to all forms of abnormal gonads, which can occur in individuals with normal karyotypes (46,XX; 46,XY) as well as a variety of abnormal or mosaic states, most commonly Turner’s syndrome (45,XO). The gonads are usually streaks of fibrous tissue.
Mixed Gonadal Dysgenesis
Patients who present with gonadal dysgenesis and a normal karyotype need to be assessed for a variety of other conditions, such as neurosensory deafness and fragile X syndrome. These clinical associations are particularly true when familial premature ovarian failure is identified.9
General Principles of X Chromosome Genetic Disorders
Translocations of the X chromosome, although extremely rare, may cause amenorrhea depending on the location of breakpoints. In a balanced X translocation one X chromosome is normal, and the other is an X autosome translocation chromosome. X inactivation is not usually random so that the normal X is usually inactivated. If the translocated chromosome were inactivated, the autosome would also be inactivated, making the karyotype lethal. Nearly all males and half of the females with X autosome translocations are sterile.10
Turner’s Syndrome
It is known that specific genes in the X chromosome are essential for normal functioning of the ovaries.11 It appears that both X chromosomes with normally functioning genes need to be present in the oocytes to prevent the formation of a streak gonad.
The characteristic physical features common to females with Turner’s syndrome include short stature, somatic abnormalities (webbed neck, shield chest, increased angle at the elbow known as cubitus valgus, cardiovascular abnormalities), and prepubertal status associated with elevated gonadotropins.12 Patients with Turner’s syndrome require special attention to the autoimmune disorders and renal anomalies that are frequently found with the condition. All patients with Turner’s syndrome should seek expert cardiology consultation and screening, including chest X-ray and echocardiography, at the time of diagnosis. Annual cardiac examinations, including evaluation of blood pressure, and repeated screening at 3- to 5-year intervals if the initial screening reveals no abnormalities. When the cardiac echo is abnormal or the ascending aorta cannot be visualized, magnetic resonance imaging (MRI) of the chest should be performed in all patients.12
Gonadal Dysgenesis 46,XY: Swyer Syndrome
About 10% to 15% of patients with Swyer syndrome possess mutations of the SRY (sex-determining region on the Y chromosome) gene located on the distal portion of Yp.13,14 Due to the significant increased risk for tumor development in patients with a Y chromosome and streak gonads, gonadectomy is recommended at an early age.
Management of Gonadal Dysgenesis
Oocyte donation offers women with Turner’s syndrome the opportunity to achieve pregnancy. However, the increased cardiovascular demands of pregnancy also may pose unique and serious risk given their high rate of cardiovascular malformations (25% to 50% prevalence). A recent Practice Committee position from the American Society for Reproductive Medicine (ASRM) has stated that because the risk for aortic dissection or rupture during pregnancy may be 2% or higher, the risk of death during pregnancy is increased as much as 100-fold.12 Any significant cardiac abnormality should be regarded as a contraindication to oocyte donation. Even those having a normal evaluation should be thoroughly counseled regarding the high risk of cardiac complications during pregnancy because aortic dissection may still occur.12
Disorders of the Genital Tract
Müllerian Agenesis
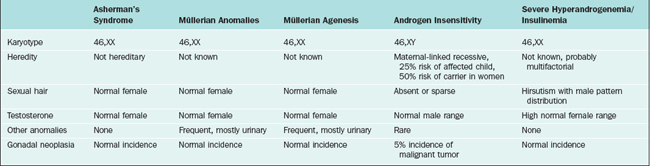
Mullerian agenesis, also referred to as Mayer-Rokitansky-Küster-Hauser syndrome, is a condition in which all or part of the uterus and vagina are absent in the presence of otherwise normal female sexual characteristics. This diagnosis accounts for approximately 10% of cases associated with primary amenorrhea.15 In Finland, the incidence was calculated to be approximately 1 of every 5000 newborn girls.16 In müllerian agenesis, the ovaries are not affected and thus ovarian function is normal. Secondary sexual development and height are in a normal range.
The differential diagnosis of patients who present with primary amenorrhea and have a genital tract anomaly is summarized in Table 16-4. Partial development of the müllerian structures can lead to obstructed menses and painful distention of a hematocolpos, hematometra, or hematoperitoneum. It is important to separate müllerian agenesis from complete androgen insensitivity syndrome, because the vagina may be absent or short in both disorders.17
It is currently unknown why müllerian agenesis occurs, but likely causes are mutations of genes that are responsible for müllerian tract maintenance. Thus far, no mutations have been reported.18 It appears that the mode of inheritance is not autosomal dominant.19
Although ultrasound could be an important aid in confirming the presence or absence of uterine structures, MRI is usually more definitive. Occasionally laparoscopic visualization is needed, because there is disagreement between MRI and laparoscopic findings.20 If persistent chronic pelvic pain and symptoms associated with endometriosis are present, laparoscopy can usually aid in determining the location and potential removal of incompletely formed müllerian structures.
Outflow Obstructions
Imperforate hymen is the most frequent obstructive female genital tract anomaly, with an estimated frequency of approximately 0.1%. Imperforate hymen usually occurs sporadically, but familial cases have been reported.21 Although this entity can present in infants or children of any age as a mucocolpos, a bulging intact hymen with hematocolpos presenting at the time of menarche is not unusual. Hymenectomy effectively alleviates this problem.
A transverse vaginal septum is somewhat less common than an imperforate hymen, occurring in fewer than 1 in 20,000 females. The presentation and surgical treatment is similar to an imperforate hymen if the transverse vaginal septa is located in the lower third of the vagina. However, more than 80% are located in the middle and upper vagina. Diagnosis is usually made by ultrasound or MRI. When located in these areas, septa tend to be thicker, and surgery is more difficult. Surgical management is described in detail in Chapter 51.
Androgen Insensitivity Syndrome
Androgen insensitivity syndrome (formerly known as testicular feminization), is an X-linked recessive condition in which genotypic males (46,XY) develop into apparently phenotypic females who have no müllerian structures and intra-abdominal testes. The underlying cause is an abnormally functioning androgen receptor, which prevents normal masculinization. This syndrome will be identified in as many as 5% of all patients presenting with primary amenorrhea.17
Evaluation of Androgen Insensitivity Syndrome
On pelvic examination, the patients will have scant or absent pubic hair and a blind vagina that is no more than a few centimeters long. More than 50% of patients have inguinal hernias and underdeveloped labia minora. Laboratory evaluation will demonstrate a 46,XY karyotype and elevated total testosterone (in the normal male range).22 This distinguishes patients with androgen insensitivity syndrome from those with müllerian agenesis, who will have an XX karyotype and total testosterone in the normal female range.
Rare Causes of Primary Amenorrhea
Isolated Gonadotropin Deficiency
This disorder is characterized by a decrease or absence in endogenous gonadotropin-releasing hormone (GnRH) secretion, which results in very low to undetectable luteinizing hormone (LH) and FSH levels. Individuals with the disorder have incomplete development of secondary sexual characteristics, primary amenorrhea, eunuchoid features, and in some cases a decreased sense of smell or anosmia (Kallmann syndrome). Male patients with Kallmann syndrome have X-linked recessive idiopathic hypogonadotropic hypogonadism accompanied by anosmia caused by mutations in the KAL1 gene, localized to the pseudoautosomal region of Xp.23,24
The protein product of KAL1, anosmin, possesses neural cell adhesion molecule properties. Anosmin provides a guide for GnRH neurons and olfactory nerves to migrate from the olfactory placode across to the olfactory bulb. When anosmin is absent or defective, GnRH and olfactory neurons fail to synapse normally. The KAL1 gene escapes X inactivation and an inactive pseudogene is present on Yq. For individuals with anosmia or hyposmia, there is evidence of hypoplasia of the olfactory bulbs on MRI. No KAL1 gene mutations have been identified in females with idiopathic hypogonadotropic hypogonadism and anosmia, suggesting that other autosomal genes may be involved.25
LH Receptor Abnormalities
Abnormalities of the LH receptor in 46,XX females will result in normal female sexual development and primary amenorrhea.26 Serum LH may be normal to increased, FSH is normal, follicular phase estradiol levels are normal, and progesterone is low. The uterus is small and the ovaries are consistent with anovulation.
Gonadotropin-Releasing Hormone Receptor Abnormalities
Abnormalities of GnRH receptors have also been identified. The phenotype of GnRH resistance ranges from complete idiopathic hypogonadotropic hypogonadism to oligo-ovulation. Baseline levels of LH and FSH may be in the prepubertal or normal range. However, levels of other pituitary hormones, such as thyrotropin, growth hormone, prolactin, and corticotropin, are normal. Due to the failure to increase gonadal sex steroid secretion during puberty, secondary sex characteristics fail to develop and closure of the epiphyseal plates of the long bones is delayed, resulting in a eunuchoid habitus in which the arm span is greater than the height. Even in patients with complete forms, pulsatile GnRH administration may increase pituitary gonadotropin response; pregnancies have been reported.27
Diagnostic Approach for Primary Amenorrhea
What Age?
An evaluation of primary amenorrhea is indicated when an adolescent fails to menstruate by age 15 in the presence of normal secondary sexual development (two standard deviations above the mean of 13 years), or within 5 years after breast development, if that occurred before age 10.2 This age has recently been adjusted to a younger age, because girls are menstruating at a younger age. If there is a failure to initiate breast development by age 13 (two standard deviations above the mean of 10 years), an investigation should also be initiated.2 These criteria are not absolute, and complete evaluation should be initiated if the patient presents with amenorrhea and obvious associated pathology such as cyclic pain or a blind vaginal pouch.
History and Physical Examination
A careful history is a key component to the evaluation and planning for the treatment of amenorrhea (Fig. 16-1). Special emphasis should be focused on physical or emotional stress, nutritional status, and history of genetic inherited disorders. The family should be questioned about familial disorders such as diabetes mellitus, previous medical disorders that may have been treated with gonadotoxic agents, and surgical disorders that involve the genital tract, including abnormal sexual differentiation. The functional inquiry should include secondary sexual development, galactorrhea, and the presence of hyperandrogenic symptoms.
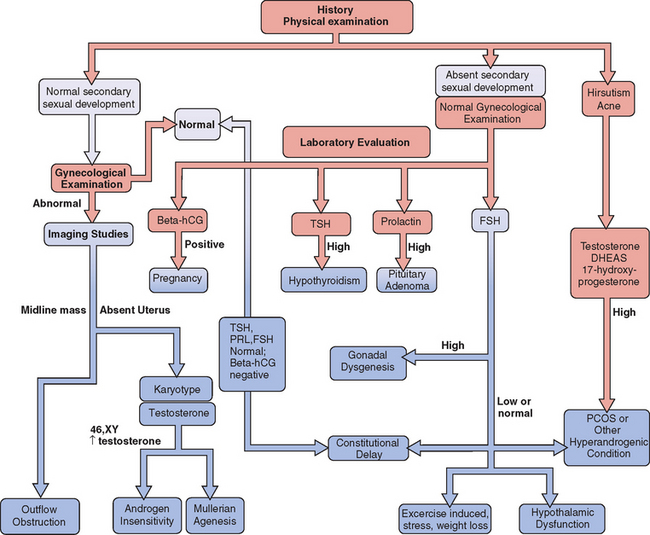
Figure 16-1 Flow diagram in the evaluation of adolescents with primary amenorrhea, showing major decision points.
Imaging
In patients with primary amenorrhea, physical examination will often detect genital tract anomalies. However, the characterization of the specific anomaly often requires imaging studies. In some cases, abdominal ultrasonography is adequate to determine the presence or absence of a uterus. Probably the most effective imaging method for characterizing congenital anomalies is MRI of the pelvis. Congenital anomalies are considered in depth in Chapters 12 and 51.
Laboratory Evaluation
If hirsutism is present or if androgen insensitivity syndrome is suspected, androgens should be evaluated. Total testosterone will be elevated in this syndrome as well as in cases of ovarian tumors. Dehydroepiandrosterone sulfate (DHEAS) will be elevated in patients with adrenal tumors and Cushing’s disease. Borderline elevation of these androgens is often seen with polycycstic ovary syndrome. 17-hydroxyprogesterone is often elevated in patients with adult onset congenital adrenal hyperplasia, although a provocative test is often necessary in subtle cases.






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


