CHAPTER 84 Alcoholic Liver Disease
EPIDEMIOLOGY
Although two thirds of American adults drink alcohol, only a minority are problem drinkers. Nevertheless, the number of alcoholics in the United States is estimated to be 14 million.1 The total costs of alcohol abuse amount to $185 billion annually, most of which are related to lost productivity and motor vehicle collisions. Alcoholic liver disease also is a major health care problem, accounting for 40% of deaths from cirrhosis and more than 30% of cases of hepatocellular carcinoma in the United States.1,2 In both Europe and the United States, alcoholic liver disease and its complications account for 50,000 deaths annually.3
Numerous studies have shown that alcoholic liver disease develops in women after a shorter duration of drinking and with a lower daily alcohol intake than in men.4,5 Population-based surveys have documented that men usually must drink 40 to 80 g of alcohol daily and women 20 to 40 g daily for 10 to 12 years to achieve a significant risk of liver disease.4–6 Table 84-1 illustrates the alcohol content of various beverages, their typical serving sizes, and the daily alcohol intake, for at least 10 years, that puts both men and women at risk for the development of alcoholic liver disease.

SPECTRUM OF DISEASE
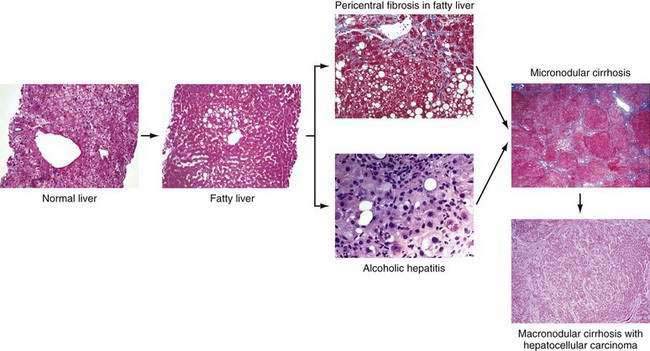
Chronic alcohol abuse can result in a spectrum of liver injury that ranges from mild fatty infiltration to cirrhosis and hepatocellular carcinoma (Fig. 84-1).7,8 Fat accumulation in liver cells, which is the earliest and most predictable response to alcohol ingestion, is seen in 90% of heavy drinkers. Although fatty liver generally is a benign condition that usually reverses quickly with abstinence, cirrhosis can develop within five years in 10% of patients who continue to drink heavily.9 Cirrhosis is particularly likely to develop if the steatosis manifests as a mixed micro- and macrovesicular pattern rather than the macrovesicular pattern seen in most alcoholics.8 Much more important than steatosis alone is the development of necroinflammation, with or without fat infiltration, and fibrosis (alcoholic hepatitis) that occurs in approximately 10% to 35% of heavy drinkers. Alcoholic hepatitis is an important clinical entity for the following two reasons: (1) patients with severe alcoholic hepatitis have extremely high short-term mortality rates, and (2) alcoholic hepatitis is a well-documented precursor of cirrhosis, with an associated long-term risk of cirrhosis that is nine times higher than that for patients with fatty liver alone.9 With continued alcohol abuse, a fine mesh-like pattern of cirrhosis with prominent involvement of the central vein (micronodular, or Laennec’s, cirrhosis) develops in 8% to 20% of heavy drinkers. Over time, and especially with continued abstinence, this lesion can evolve to include broad bands of fibrosis that separate large nodules of liver tissue (macronodular cirrhosis). Hepatocellular carcinoma typically develops in this setting.
PATHOGENESIS
ETHANOL METABOLISM AND TOXIC METABOLITES
The liver is the main organ responsible for ethanol metabolism; other organs such as the stomach contribute to much lesser degrees. Ethanol is metabolized by three major systems in the liver: alcohol dehydrogenase (ADH), cytochrome P450 2E1 (CYP2E1), and, of least importance, catalase.10 ADH is actually a set of cytoplasmic enzymes with multiple isoforms. ADH is the primary enzyme system responsible for metabolism of ethanol at low concentrations, whereas CYP2E1 contributes at higher concentrations of ethanol (greater than 10 mM) and is induced by exposure to ethanol. Both ADH and CYP2E1 convert ethanol to acetaldehyde, which is then converted to acetate by aldehyde dehydrogenase (ALDH). Acetaldehyde is a highly reactive and potentially toxic compound that is responsible for many of the systemic toxic effects of alcohol, such as nausea, headaches, and flushing. The “Oriental flush syndrome” results from impaired metabolism of acetaldehyde caused by inheritance of the ALDH22 allele, which encodes an inactive form of ALDH2. Persons from East Asia who are homozygous for this mutation rarely drink ethanol because they invariably experience toxic systemic effects, such as flushing and tachycardia, when they do drink.
Acetaldehyde also is postulated to play an etiologic role in alcoholic liver disease. Acetaldehyde can form adducts with reactive residues on proteins or small molecules (e.g., cysteines). These chemical modifications can alter or interfere with normal biologic processes and can be directly toxic to the cell. Modified molecules also may stimulate the host’s immune response and cause autoimmune-like manifestations. Antibodies against such oxidatively modified proteins have been reported in both human and animal models of alcoholic liver disease.11 An example is the hybrid adduct of malondialdehyde and acetaldehyde (MAA), unique to alcohol exposure, which induces an immune reaction in human alcoholics and in animal models of alcoholic liver disease.11 Acetaldehyde also has been shown to impair mitochondrial glutathione transport and to sensitize hepatocytes to tumor necrosis factor (TNF)-mediated killing.12
In addition to forming cytotoxic metabolites such as acetaldehyde, ethanol metabolism can alter the cellular oxidation-reduction (redox) state, thereby modulating liver injury. Specifically, the oxidation of ethanol uses nicotinamide-adenine dinucleotide (NAD+) as an electron acceptor and thereby causes a shift in the ratio of reduced NAD (NADH) to NAD+ to a more reduced state.10 This change in the redox state can impair normal carbohydrate and lipid metabolism; multiple effects ensue, including a decrease in the supply of adenosine triphosphate (ATP) to the cell and an increase in hepatic steatosis.
OTHER METABOLIC MECHANISMS
Oxidative Stress
Oxidative stress is an imbalance between pro-oxidants and antioxidants. Reactive oxygen species (ROS) and reactive nitrogen species (RNS) are products of normal metabolism and can be beneficial to the host (e.g., by contributing to bacterial killing).13 Overproduction of ROS and RNS or inadequate antioxidant defenses (e.g., low levels of vitamins, selenium, mitochondrial glutathione), or both, can lead to liver injury. Oxidative stress is well documented in alcoholic liver disease.14 Studies in normal volunteers have shown that acute alcohol consumption causes a dose-related increase in urinary isoprostane levels (a marker of lipid peroxidation, which is an indirect marker of oxidative stress), and patients with alcoholic hepatitis exhibit high isoprostane levels.15 Major ROS and RNS include superoxide anion, hydrogen peroxide, and hydroxyl radical (ROS) and nitric oxide, peroxynitrite (RNS), and hypohalous acid. Oxidative stress usually is documented by detection of one of several indirect markers (1) protein oxidation (e.g., protein thiol or carbonyl products); (2) lipid oxidation (e.g., isoprostanes, malondialdehyde); (3) DNA oxidation (e.g., oxodeoxyguanosine); or (4) depletion or induction of antioxidant defenses (e.g., vitamin E, glutathione, thioredoxin).
The stimulus for oxidative stress in the liver comes from multiple sources. In hepatocytes, CYP2E1 activity increases after alcohol consumption—in part because of stabilization of messenger RNA (mRNA). The CYP2E1 system leaks electrons to initiate oxidative stress.13 CYP2E1 is localized in the hepatic lobule in areas of alcohol-induced liver injury. Moreover, overexpression of CYP2E1 in mice and in HepG2 cells (a hepatocyte cell line) in vitro leads to enhanced alcohol hepatotoxicity.16,17 On the other hand, alcohol-induced liver injury still develops in CYP2E1-knockout mice. These findings suggest that increased CYP2E1 probably plays a role in alcoholic liver injury but is not the sole or dominant factor. In the CYP2E1-knockout mice, other compensatory mechanisms may be operational, such as induction of other microsomal enzymes. Nonparenchymal cells and infiltrating inflammatory cells (e.g., polymorphonuclear neutrophils) are another major source of pro-oxidants that are used for normal cellular processes, such as killing invading organisms. Major enzyme systems for pro-oxidant production in Kupffer cells include nicotinamide-adenine dinucleotide phosphate (NADPH) oxidase and inducible nitric oxide synthase (iNOS). Mice deficient in NADPH oxidase or mice treated with the drug diphenyleneiodonium sulfate, which blocks NADPH oxidase, are resistant to ethanol-induced liver injury.18,19 Infiltrating neutrophils use enzyme systems such as myeloperoxidase to generate hypochlorus acid (HOCl−, a halide species that causes oxidative stress) and RNS.
The critical role of oxidative stress in the development of alcoholic liver disease has been validated in multiple studies in rats and mice fed alcohol and treated with various antioxidants, ranging from ebselen to green tea polyphenols, that overexpress both superoxide dismutase I and II. Various antioxidants have been shown to block or attenuate the development of alcoholic liver disease in rodents.13
Mitochondrial Dysfunction
Mitochondria are the major consumers of molecular oxygen and major generators of ROS in the liver. Mitochondrial dysfunction is well documented in alcoholic liver disease and contributes to oxidative stress.20,21
Mitochondrial abnormalities in alcoholic liver disease include megamitochondria observed on light and electron microscopy and functional mitochondrial abnormalities as documented by an abnormal 13C ketoacid breath test result (ketoacids are metabolized by mitochondria).22 Short-term administration of alcohol causes increased hepatic superoxide generation in liver mitochondria, with an increased flow of electrons along the respiratory electron transport chain. The increased NADH/NAD+ ratio caused by ethanol intake favors superoxide generation.13 Because hepatic mitochondria lack catalase, glutathione plays a critical role in protecting mitochondria against oxidative stress. Mitochondria do not make glutathione but instead import it from the cytosol. In alcoholic liver disease, the transport of glutathione into mitochondria is impaired, and selective mitochondrial glutathione depletion is observed.23 Glutathione depletion also sensitizes the liver to the toxic effects of TNF, and TNF also impairs mitochondrial function. Finally, an increase in mitochondrial membrane depolarization and permeability leads to hepatocyte death, especially apoptotic (programmed), rather than necrotic, cell death.
Hypoxia
The centrilobular area of the hepatic lobule (the functional unit of the liver) has the lowest oxygen tension and greatest susceptibility to hypoxia. Chronic alcohol intake increases oxygen uptake by the liver and increases the lobular oxygen gradient. A chronic intragastric feeding model in rats has been used to define the mechanisms underlying hepatic hypoxia and the association of these mechanisms with cycling of urinary alcohol levels (UALs).24 At high UALs, hepatic hypoxia is observed, along with reduced ATP levels; the NADH/NAD+ ratio is shifted to the reduced state; and the hypoxia-inducible factor (HIF) 1 and 2 genes are up-regulated. When UALs fall, reperfusion injury occurs, with free radical formation and peak liver enzyme release from hepatocytes. Stimuli for this cycle of events include catecholamines (levels of which coincide with peak UALs) and modulators of vascular tone (e.g., nitric oxide, endothelin-1); modulation of these stimuli offers future therapeutic possibilities.
Impaired Proteasome Function
The 26S ubiquitin-proteasome pathway is the primary proteolytic pathway of eukaryotic cells (see Chapter 72). It controls the levels of numerous proteins involved in gene regulation, cell division, and surface receptor expression, as well as stress response and inflammation. The proteasome system is now considered a cellular defense mechanism because it also removes irregular and damaged proteins generated by mutations, translational errors, or oxidative stress.25 This pathway involves two major steps: (1) covalent attachment of multiple ubiquitin molecules to the protein substrate and (2) degradation of the targeted protein by the 26S proteasome complex. The 26S ubiquitin-proteasome pathway may play a pathogenic role in the development of alcoholic liver disease.26
Early clinical studies showed that hepatomegaly caused by chronic alcohol consumption resulted in part from accumulation of protein in the liver.27 Animal studies have demonstrated that chronic ethanol feeding results in a significant decrease in proteolytic activity of the proteasome; this decreased activity can lead to abnormal protein accumulation, including accumulation of oxidized proteins.28,29 The decrease in proteasome function correlates significantly with the level of hepatic oxidative stress. Patients with alcoholic cirrhosis have increased serum ubiquitin levels, suggesting damaged proteasome function.30 Also, hepatocytes from alcoholics contain large amounts of ubiquitin in the form of cellular inclusions, or Mallory (or Mallory-Denk) bodies, which accumulate because they are not degraded efficiently by the proteasome.31 The formation of Mallory bodies, which occurs when the ubiquitin-proteasome system is inhibited or overwhelmed, is a pathway of liver injury caused by diverse toxins, including alcohol.31 As hepatocytes die as a result of proteasome inhibition, they inappropriately release cytokines such as interleukin (IL)-8 and IL-18. IL-8 recruits neutrophils and probably plays a role in neutrophil infiltration in alcoholic hepatitis, and IL-18 sustains inflammation in the liver.32
Abnormal Metabolism of Methionine, S-adenosylmethionine, and Folate
In mammals, the liver plays a central role in methionine metabolism; nearly one half of the daily intake of methionine is metabolized in the liver (Fig. 84-2). The first step in methionine metabolism is the formation of S-adenosylmethionine (SAM) in a reaction catalyzed by methionine adenosyltransferase (MAT). Activity of this enzyme is depressed in alcoholic liver disease.33 SAM is the principal biological methyl donor through the transmethylation pathway; the precursor of aminopropyl groups used in polyamine biosynthesis; and a precursor of glutathione through its conversion to cysteine along the transsulfuration pathway. Under normal conditions, most of the SAM generated daily is used in transmethylation reactions, in which methyl groups are added to a large number of molecules by means of specific methyltransferases.34 These compounds include DNA, RNA, biogenic amines, phospholipids, histones, and other proteins; methylation of these compounds may modulate cellular functions and integrity. In this process, SAM is converted to S-adenosylhomocysteine (SAH), which is a potent competitive inhibitor of most methyltransferases. Both an increase in SAH and a decrease in the SAM/SAH ratio are known to inhibit transmethylation reactions.34–36
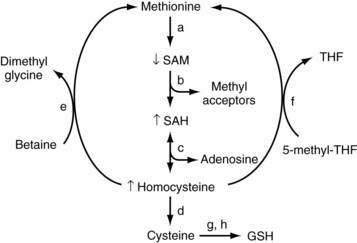
Deficiency of SAM in patients with alcoholic liver disease was first noted in the early 1980s, when it was observed that alcoholic subjects had delayed clearance of an oral bolus of methionine (presumably because of blocked conversion of methionine to SAM).37 Functional MAT activity was subsequently shown to be subnormal in liver biopsy specimens from alcoholic subjects.38 Subnormal hepatic SAM levels also are noted in various experimental models of liver injury. Exogenous administration of SAM corrects the deficiency and attenuates the severity of these experimental forms of liver injury.33,39
Because SAM is a precursor of glutathione, SAM deficiency results in glutathione deficiency, which is observed in many forms of liver disease.40 In animal studies, exogenous SAM corrects hepatic deficiencies of both SAM and glutathione.41 Because glutathione is required for optimal expression of MAT activity in liver, hepatic deficiency of MAT may be caused in part by glutathione deficiency. Also, hepatic MAT is sensitive to oxidative stress, and the subnormal hepatic MAT activity in patients with alcoholic liver disease could result from oxidation of MAT.42
In models of alcohol-induced hepatotoxicity, SAM has been shown to maintain mitochondrial glutathione levels. Depletion of mitochondrial glutathione is thought to be one pathogenic factor in the development of alcoholic liver disease, and SAM, but not other glutathione prodrugs, prevents mitochondrial glutathione depletion in experimental alcoholic liver disease (possibly by protecting mitochondrial glutathione transport systems).43 SAM also decreases lipopolysaccharide (LPS)-stimulated TNF release and increases IL-10 release in a monocyte cell line.33 Similarly, in rats fed a diet to induce SAM deficiency, serum TNF levels increase, and sensitivity to endotoxin-induced hepatotoxicity, which can be blocked by injection of SAM, increases markedly.44 These data support the concept that SAM may have direct hepatoprotective functions and may modify LPS-stimulated cytokine production.
Although serum SAM levels are decreased in patients with alcoholic liver disease, levels of the downstream products SAH and homocysteine are elevated. Homocysteine has been postulated to play a role in the pathogenesis of fatty liver seen with alcoholic liver disease, and in experimental animals reduction of homocysteine levels with administration of betaine (to convert homocysteine to methionine) attenuates the severity of alcoholic liver disease.45 Elevated SAH levels have been shown to sensitize hepatocytes to TNF-mediated destruction, and SAH may be a critical physiologic sensitizer of TNF-mediated killing in liver injury.36 Homocysteine and SAH can be removed by giving betaine, which facilitates regeneration of methionine from homocysteine. Folic acid also can play a critical role in the regeneration of homocysteine to methionine by means of 5-methyltetrahydrofolate (5-MTHF).46 Fatty liver develops in mice that lack the gene for MTHF reductase (MTHFR), and steatohepatitis develops in MAT1A–knockout mice; these findings further support a role for this critical pathway in the development of steatosis and steatohepatitis. Halsted and colleagues have shown that folic acid deficiency enhances the development of alcohol-induced liver injury in micropigs and that alcohol feeding interferes with normal folic acid metabolism in multiple different pathways—from impaired intestinal uptake to increased renal excretion.46 Collectively, the data support a role for altered methionine-transmethylation-transsulfuration metabolism in alcoholic liver disease and link these pathways to TNF hepatotoxicity.35
IMMUNE AND INFLAMMATORY MECHANISMS
Kupffer Cell Activation and Dysregulated Cytokine Production
Cytokines are low-molecular-weight mediators of cellular communication (see Chapter 2).47 Multiple cell types in the liver are potential sources of the increased release of proinflammatory cytokines observed in alcoholic liver disease. Kupffer cells are prominent producers of proinflammatory cytokines such as TNF-α, as well as certain anti-inflammatory cytokines such as IL-10. Sinusoidal endothelial cells express adhesion molecules, which modulate white blood cell adhesion and transmigration. Activated stellate cells produce collagen in response to signals such as the profibrotic cytokine transforming growth factor-β (TGF-β). Hepatocytes are a relatively newly recognized source of cytokine production, including IL-8, a major neutrophil chemotactic peptide and angiogenesis factor.
Major stimuli for the observed increase in proinflammatory cytokine production in alcoholic liver disease are believed to be ROS and intestine-derived LPS.47 In alcoholic liver disease, intestinal permeability is increased and the frequency of endotoxemia is high. LPS activates the redox-sensitive transcription factor NF-κB in Kupffer cells, thereby resulting in the production of certain cytokines such as TNF (Fig. 84-3). TNF can increase intestinal permeability, induce oxidative stress, and perpetuate this cycle. Generation of ROS through the metabolism of alcohol also can activate NF-κB and stimulate proinflammatory cytokine production.48 Although necrosis has traditionally been thought to be the major mechanism of hepatocyte cell death in alcoholic liver disease, increased apoptosis also has been documented. As hepatocytes die of apoptosis, they can be taken up by Kupffer cells and stimulate TNF production.49 Moreover, when alcohol-fed rodents are treated with caspase inhibitors to block apoptosis, liver inflammation and injury are markedly attenuated.50 When hepatocytes die of proteasome inhibition-mediated apoptosis, the dying hepatocytes release IL-8 and IL-18, which cause sustained inflammation.32 Therefore, in alcoholic liver disease, hepatocyte apoptosis may sustain proinflammatory cytokine production and cell injury or death.
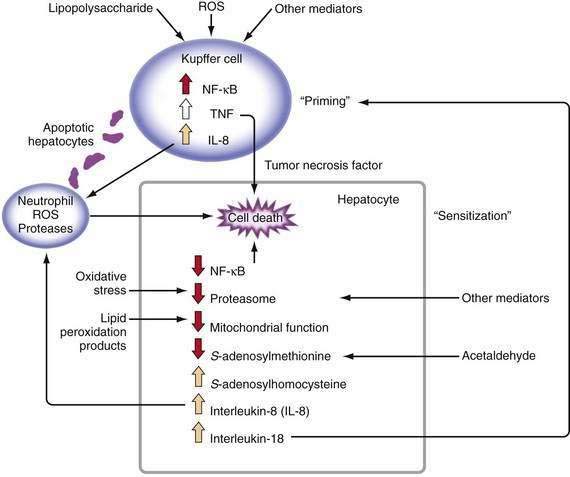
TNF metabolism is dysregulated in alcoholic hepatitis, as suggested by the observation that cultured monocytes (which produce substantial amounts of TNF) from patients with alcoholic hepatitis produce significantly increased amounts of TNF in response to LPS stimulation.51 Increased serum TNF concentrations in patients with alcoholic hepatitis show a strong correlation with disease severity and risk of mortality.47 Serum concentrations of TNF-inducible cytokines and chemokines, such as IL-6, IL-8, IL-18, monocyte chemoattractant protein 1 (MCP-1), and others, are elevated in patients with alcoholic hepatitis or cirrhosis, and the levels often correlate with markers of the acute-phase response, reduced liver function, and poor clinical outcomes.47
This enhanced cytokine response to a physiologic stimulus such as LPS is termed priming. Increased serum or urinary levels of neopterin and other markers indicate that monocytes and Kupffer cells are primed in alcoholic liver disease. This priming for LPS-stimulated TNF production has been reproduced in vitro by culturing monocyte cell lines with relevant concentrations of alcohol. This response appears to be mediated, at least in part, by induction of CYP2E1 and oxidative stress.52
Studies in rats, mice, and tissue culture have validated a pathogenic role for cytokines (especially TNF) in the development of alcoholic liver disease.47 Rats chronically fed alcohol are more sensitive to the hepatotoxic effects of injected LPS and have much higher LPS-stimulated plasma levels of TNF in comparison with control rats. Liver injury can be attenuated by giving a prostaglandin analog to down-regulate TNF production.53 Because rats have a natural aversion to alcohol, an intragastric route is often used to deliver high amounts of alcohol. Studies using the intragastric alcohol-feeding model have demonstrated that the development of liver injury coincides with an increase in TNF mRNA expression in the liver and in isolated Kupffer cells.54,55 Rats fed ethanol intragastrically also have high blood LPS levels and increased expression of CYP2E1, as well as markers of oxidative stress and lipid peroxidation.
Not only are levels of proinflammatory cytotoxic cytokines increased in alcoholic liver disease, but also monocyte and Kupffer cell production of protective anti-inflammatory cytokines such as IL-10 is decreased.56 The importance of this observation for humans has been confirmed using IL-10–knockout mice, in which more severe ethanol-induced hepatotoxicity develops and increased levels of pro-inflammatory cytokines such as TNF are seen.57
Several strategies have been devised to decrease cytokine production or activity in an attempt to block or attenuate liver injury. Examples include antibiotics to modulate intestinal flora and LPS, gadolinium chloride to destroy Kupffer cells, and antioxidants such as glutathione prodrugs to inhibit cytokine production. Each of these strategies has been successful in attenuating alcohol-induced liver injury in rats.47 Prebiotics such as oat bran also have been shown to decrease endotoxemia in experimentally induced alcoholic liver injury. Perhaps the most compelling data that relate TNF to alcohol-induced liver injury are from studies in which anti-TNF antibody has been used to prevent liver injury in alcohol-fed rats.58 Similarly, alcoholic liver injury does not develop in mice that lack the TNF type I receptor.59
Hepatocytes normally are resistant to TNF killing. Hepatocytes from rats fed alcohol or hepatocytes incubated in alcohol are sensitized to TNF killing, however.16,60 Some potentially relevant mechanisms for this sensitization include mitochondrial depletion of glutathione, accumulation of SAH, and inhibition of proteasomes, among others. Therefore, in alcoholic liver disease, monocytes and Kupffer cells are primed to increase production of TNF and to sensitize hepatocytes to TNF killing. These processes are closely intertwined with previously described mechanisms such as oxidative stress, mitochondrial dysfunction, abnormal metabolism of methionine, and dysfunction of proteasomes.
Immune Responses to Altered Hepatocellular Proteins
Alcoholic hepatitis may persist histologically for many months after exposure to ethanol has ceased, suggesting an ongoing immune or autoimmune response. Autoimmune reactions are now well documented in patients with alcoholic liver disease, with autoantibodies directed against phospholipids, alcohol dehydrogenase, heat shock protein, and other potential antigens.61 Patients with alcoholic liver disease are at increased risk for the development of immune responses directed at neoantigens generated from the interactions of metabolites of alcohol (e.g., acetaldehyde or hydroxyethyl radicals) with liver proteins.62 Studies have linked genetic susceptibility and autoimmunity in alcoholic liver disease. Some humans have a genetic mutation in the cytotoxic T lymphocyte–associated antigen 4G (CTLA-4G) allele that leads to inappropriately activated T cell function. One of the breakdown products of alcohol metabolism, the hydroxyethyl radical, can modify CYP2E1 and, in the presence of the CTLA-4G mutation, increase the risk that anti-CYP2E1 autoantibodies will develop.63 This is one pathway whereby alcohol abuse may break “self-tolerance” in the liver.
GENDER AND GENETIC FACTORS
Female gender is now a well-accepted risk factor for the development and rapid progression of alcoholic liver disease.4,5 Although rates of metabolism and elimination of alcohol have been reported to be more rapid in women than in men, when adjusted for liver volume, elimination rates are similar between genders.64 Studies in rats or mice fed alcohol chronically have demonstrated that females are more susceptible than males to liver injury. Risk factors for the development of liver disease in females appear to include increased endotoxemia, lipid peroxidation, and chemokine (e.g., monocyte chemotactic protein-1) mRNA levels and activation of the critical transcription factor NF-κB.65 These risk factors are critical for determining “safe” levels of alcohol consumption in women. Many authorities consider any amount of alcohol above 20 g a day to be a risk factor for the development of liver disease in women; however, differences in levels of alcohol dehydrogenase in gastric mucosa between men and women are not thought to play a major role in the greater susceptibility of women to alcoholic liver injury.
Genetic polymorphisms in alcohol-metabolizing systems such as CYP2E1 and ADH have been postulated to play a role in the development of alcoholic liver disease.66 None of these polymorphisms, however, adequately explains the diverse pathologic responses seen among patients with alcoholic liver disease. Polymorphisms in the promoter regions of cytokines TNF and IL-10 also have been reported to predispose affected persons to the development of alcoholic liver disease and are under active study.67,68
EMERGING MECHANISMS
Three areas deserve recognition as emerging mechanisms of liver injury. The first is the area of epigenetics, which refers to changes in gene expression that do not involve DNA coding sequence modifications. Epigenetic mechanisms typically involve DNA methylation and a variety of modifications of histones. Epigenetic modifications can be transient or long term and can be tissue or organ specific. The critical steps and enzymes (e.g., histone acetyltransferase, DNA methyltransferase) responsible for epigenetic modifications are under active investigation by multiple laboratories.69
A second area is the endoplasmic reticulum (ER) stress response, which is induced by the accumulation of unfolded or misfolded proteins. To deal with the ER stress response, cells activate a series of signaling pathways termed the unfolded protein response (UPR), which can be either protective (usually in the short term) or detrimental (usually in the long term). One of the effects of a prolonged UPR can be increased production of triglycerides and cholesterol, leading to fatty liver. Some potential inducers of the ER stress in alcoholic liver disease include elevated homocysteine levels, acetaldehyde adducts, and oxidative stress.70
A third area is the endogenous cannabinoids, which are ubiquitous lipid signaling molecules that mediate their effects by specific cannabinoid receptors, CB1 and CB2. Studies have demonstrated that inhibition of CB1 receptors can cause weight loss and attenuate fatty liver and hyperlipidemia in animal models of obesity and steatohepatitis. Moreover, CB1 blockade reduces hepatic fibrosis in a variety of animal models of cirrhosis.71
FIBROSIS
The development of hepatic fibrosis, leading to cirrhosis, indicates major progression of alcoholic liver disease and represents a maladaptive wound healing response. The development of fibrosis is a dynamic state, with constant remodeling of scar tissue; fibrosis may regress with discontinuation of exposure to alcohol. The stellate cell is the major source of collagen production in the liver. It normally exists in a quiescent state and serves as a major storehouse for vitamin A. Following activation, the stellate cell assumes a myofibroblast-like contractile phenotype and produces collagen. The cytokine TGF-β is a major stimulus for stellate cell activation and collagen production. Other cytokines implicated in activation of stellate cells include platelet-derived growth factor and connective tissue growth factor (see Chapter 90). Oxidative stress plays a major role in stellate cell activation, and a variety of antioxidants can block both stellate cell activation and collagen production in vitro. Serum levels of 4-hydroxy-nonenal, a specific product of lipid peroxidation, are elevated in patients with alcoholic liver disease and up-regulate both procollagen type I and tissue inhibitor of metalloproteinase-1 (TIMP-1) gene expression. Matrix metalloproteinase-1 plays a major role in degrading type I collagen. TIMP-1 levels also are elevated in alcoholic liver disease. The result appears to be an increase in stellate cell activation and collagen production on the one hand and a decrease in matrix degradation on the other hand.72–74
DIAGNOSIS OF ALCOHOL ABUSE
The diagnosis of alcohol abuse is based on a history of heavy alcohol intake and the presence of other organ system damage or an excessive frequency of falls, lacerations, and fractures. Physicians typically identify only 50% of patients with drinking problems because of both inadequate questioning by physicians and denial by patients.75 Underdiagnosis is particularly common in older patients.76 The uniform application of screening tools such as the four-item AUDIT-C (Alcohol Use Disorders Identification Test) consumption questions and the four-item CAGE (need to cut down, annoyed by criticism, guilty about drinking, need for an eye-opener in the morning) questionnaire dramatically improves the recognition of patients with problem drinking in primary care clinics.77 Even the use of a single question: “When was the last time you had more than x drinks in one day?,” where x equals four for women and five for men, dramatically improves the diagnosis of alcoholism in primary care settings.78
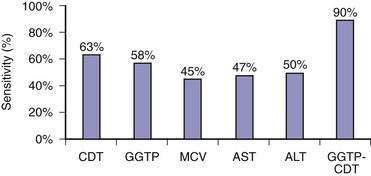
Because of the reluctance of many patients to share their drinking histories candidly, continued interest has been directed toward laboratory measures that can reliably identify patients with problem drinking. Blood or breath alcohol measurements are the most sensitive and specific indicators of recent alcohol abuse, particularly among binge drinkers.79 The major limitation of these tests is the short half-life of ethanol in blood, urine, and breath. As a result, continued efforts have focused on developing biomarkers of alcohol abuse that are detectable over longer periods of time. The most specific of these biomarkers is carbohydrate-deficient transferrin (CDT).80 CDT levels increase in serum with ingestion of 50 to 80 g/day of ethanol for two to three weeks and decline gradually during abstinence, with a half-life of approximately 15 days; however, the sensitivity of this test is only 35% to 40% among alcoholics who consume 100 g of ethanol daily.80 Conditions that can influence the CDT level include gender, age, body mass index, smoking, anorexia, pregnancy, and acute trauma with blood loss.80 These issues, as well as a lack of methodologic standardization, has limited the use of CDT measurement in clinical practice. Mean corpuscular erythrocyte volume (MCV), serum gamma glutamyl transpeptidase (GGTP) levels, and the ratio of mitochondrial aspartate aminotransferase (AST) to total AST (mitochondrial AST/total AST) and combinations of these markers have been touted as more accurate measures of alcohol abuse. A mathematically formulated equation incorporating serum GGTP and CDT levels appears to have the best diagnostic accuracy of any currently available assays for chronic alcohol abuse (Fig. 84-4).81 Measurement of two alcohol metabolites, phosphatidylethanol and ethyl glucuronide, also shows promise as an innovative way to detect recent alcohol use.80
DIAGNOSIS OF ALCOHOLIC LIVER DISEASE
HISTORY
Most patients with fatty liver are asymptomatic. Although patients with alcoholic hepatitis and cirrhosis may be asymptomatic, many present with a variety of complaints including anorexia, nausea and vomiting, weakness, jaundice, weight loss, abdominal pain, fever, and diarrhea.82
PHYSICAL EXAMINATION
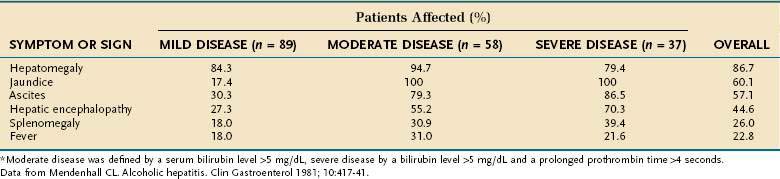
The most extensive demographic information on alcoholic liver disease in the United States comes from studies of hospitalized patients who were assigned the diagnosis on the basis of clinical and histologic parameters.82,83 Although these studies included few asymptomatic patients, they provide a useful guide to diagnosis. The most common physical finding in patients with fatty liver and alcoholic hepatitis is hepatomegaly, which is detectable in more than 75% of patients, regardless of disease severity. Patients with alcoholic hepatitis and cirrhosis also may have hepatic tenderness, an audible bruit over the liver, spider angiomata, splenomegaly, and peripheral edema. Jaundice and ascites, which are found in approximately 60% of patients, are more frequent in patients with severe disease (Table 84-2). Various degrees of hepatic encephalopathy can be seen, usually in the most severely ill patients. Some patients with alcoholic hepatitis have a fever, with temperatures as high as 104°F, that can persist for weeks.
In patients with well-compensated cirrhosis, findings on the physical examination can be normal; however, most patients have obvious hepatomegaly and splenomegaly. As the disease progresses, the liver decreases in size and has a hard and nodular consistency. Patients with decompensated cirrhosis typically have muscle wasting, ascites, spider angiomata, palmar erythema, and Dupuytren’s contractures. Enlarged parotid and lacrimal glands often are seen, and severely ill patients may have Muercke’s lines or white nails. Patients with hepatopulmonary syndrome often have digital clubbing (see Chapter 92).
LABORATORY FEATURES
Only one third of hospitalized patients with fatty liver have laboratory abnormalities, which usually consist of mild increases in serum AST and alanine aminotransferase (ALT) levels. As illustrated in Table 84-3, surprisingly modest elevations of serum aminotransferase levels are seen in patients with alcoholic hepatitis and cirrhosis, even when the disease is severe.82,83 Serum AST levels are almost always less than 300 to 500 U/L and typically are associated with trivial elevation of serum ALT levels, resulting in an AST/ALT ratio greater than 2, which is characteristic of alcoholic liver disease, in part because of deficiency of pyridoxal 5′ phosphate (a cofactor of aminotransferases) in alcoholic patients (see Chapter 73). Serum alkaline phosphatase levels can range from normal to values greater than 1000 U/L. Serum bilirubin levels range from normal to 20 to 40 mg/dL, and serum albumin levels may be normal or depressed to a value as low as 1.0 to 1.5 g/dL. Most patients with alcoholic liver disease are anemic and have some degree of thrombocytopenia. By contrast, the white blood cell count usually is normal or elevated, occasionally to levels consistent with a leukemoid state. Severely ill patients usually have marked prolongation of the prothrombin time—often expressed as the international normalized ratio (INR)—and often have elevated serum creatinine values.82–85
Related posts:
 Surgical Peritonitis and Other Diseases of the Peritoneum, Mesentery, Omentum, and Diaphragm
Surgical Peritonitis and Other Diseases of the Peritoneum, Mesentery, Omentum, and Diaphragm
 Hepatitis Caused by Other Viruses
Hepatitis Caused by Other Viruses
 Hemochromatosis
Hemochromatosis
 Gastrointestinal and Hepatic Complications of Solid Organ and Hematopoietic Cell Transplantation
Gastrointestinal and Hepatic Complications of Solid Organ and Hematopoietic Cell Transplantation
 Nutritional Assessment and Management of the Malnourished Patient
Nutritional Assessment and Management of the Malnourished Patient
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


