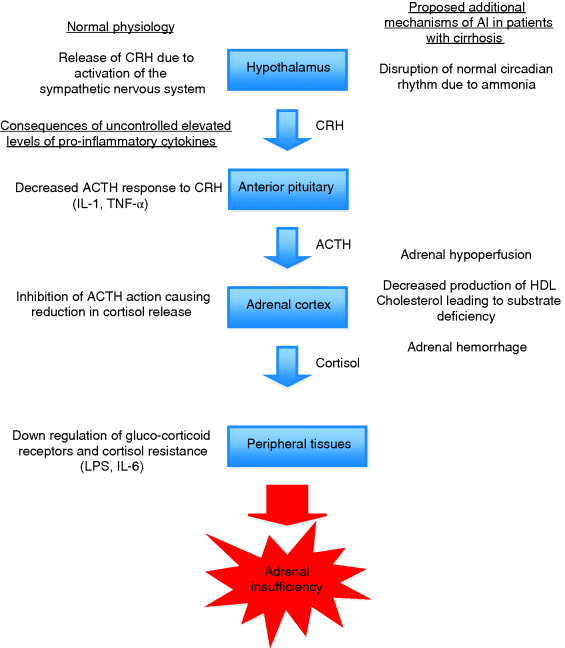
Chapter 21 Emily Dannhorn and James O’Beirne Sheila Sherlock Liver Centre, Royal Free Hospital, London, UK The concept of inadequate cortisol production during critical illness such as in septic shock has been termed relative adrenal insufficiency (RAI) and, more recently, critical illness-related corticosteroid insufficiency (CIRCI). Both these conditions have been described in critically ill patients where cortisol production and/or utilization by peripheral tissues is insufficient, leading to adverse outcomes such as increased levels of shock and mortality [1]. Due to similarities between liver failure and septic shock numerous investigators have studied the prevalence and impact of adrenal insufficiency (AI) in liver disease of various etiologies and severities. Initial results are intriguing; AI appears to be common not only in critically ill patients with liver disease, but also in patients with stable cirrhosis and post-liver transplantation. In addition, emerging evidence appears to show that AI is more prevalent in patients developing complications of cirrhosis and its presence may affect the efficacy of treatment of those complications. Currently, there is no randomized trial evidence to support the routine administration of supplemental steroids to patients with AI and complications of cirrhosis. This is partly due to difficulties encountered in the accurate measurement of cortisol in patients with cirrhosis but initial data would suggest that the presence of AI may worsen outcomes and therefore in appropriate settings the presence of AI should be considered and treated, especially if patients are not responding to conventional treatment. The hypothalamic–pituitary–adrenal axis (HPA) is instrumental in the body’s ability to deal with stress, particularly in the context of critical illness, during which cortisol has many vital roles. These include, but are not limited to, preserving vascular tone and permeability, increasing myocardial contractility, stimulating anti-inflammatory cytokine production, and initiation of lipolysis and protein catabolism [2]. Animal studies have clearly demonstrated that adrenalectomized subjects succumb rapidly to shock and corticosteroid replacement in these situations is protective [3]. In septic shock, supplemental corticosteroids have been shown to lead to faster shock reversal and low levels of serum cortisol are associated with increased mortality [4,5]. Further studies showed that some patients with septic shock had inappropriately low levels of cortisol, subnormal responses to adrenocortical stimulation, and blunted pressor responses to norepinephrine that could be restored by the administration of corticosteroids. The finding of inappropriately low cortisol levels in critically unwell patients led to the new term of relative adrenal insufficiency (RAI) [6]. More recently, the term CIRCI (critical illness-related corticosteroid insufficiency) has been used to describe a clinical picture caused by inadequate cortisol activity either by inadequate production and/or peripheral corticosteroid resistance, which is characterized by a much amplified and prolonged systemic inflammatory response [7]. Septic shock and liver failure share the same hemodynamic features (i.e., systemic vasodilatation, low mean arterial pressure, and raised cardiac output), therefore the prevalence of AI has also been studied in patients with liver disease. RAI appears to be prevalent in patients with cirrhosis and septic shock and is associated with increased mortality [8]. RAI has also been identified in patients with stable cirrhosis, nonseptic complications of cirrhosis (bleeding, ascites), and in patients post-liver transplantation [9]. The presence of AI even in the absence of critical illness in patients with cirrhosis suggests that adrenal dysfunction may be a complication of liver disease per se, leading to the term hepato-adrenal syndrome [10]. This chapter discusses the importance of AI in cirrhosis and its complications, methods of diagnosis in the correct clinical context, and treatment options. Areas where evidence is currently limited are highlighted. The body’s ability to respond appropriately to situations of stress is dependent on cortisol production by the adrenal gland regulated by the HPA axis. Under normal nonstressed conditions, cortisol secretion is pulsatile in fashion, and demonstrates marked diurnal variation. Peak levels are seen between 6 and 8 a.m. (6–23 μg/dL), with the lowest levels noted at midnight. During conditions of stress, corticotropin-releasing hormone (CRH) is released from the hypothalamus in response to inflammatory cytokines and other stimuli. CRH stimulates release of adrenocorticotropic hormone (ACTH) from the anterior pituitary, which in turn promotes the release of cortisol from the adrenal cortex. This process is tightly regulated by a series of negative feedback loops such that the release of cortisol is extremely sensitive to changes in levels of ACTH. Following its secretion, 90% of cortisol circulates in plasma bound to cortisol binding globulin (CBG), and to a lesser extent albumin. Less than 10% of cortisol circulates in the biologically active free form. Cortisol is able to diffuse rapidly across cell membranes, where it undergoes binding to glucocorticoid receptors. The glucocorticoid receptor is responsible for downstream signaling within the cell, and has several different isoforms that can be expressed differentially within various tissues, signaling different pathways [11]. Following binding to glucocorticoid receptors, the complex is translocated into the nucleus of the cell, where the effects of cortisol are exerted. Cortisol is not stored within the adrenal gland and therefore needs to be produced rapidly on demand from its precursors during times of stress. The main substrate for cortisol synthesis is cholesterol, particularly high-density lipoprotein (HDL) cholesterol. More than 80% of cortisol is produced from cholesterol at times of stress, and this is of particular relevance in conditions where cholesterol levels are low (including liver disease), where inadequate levels of substrate may limit adequate synthesis [10,12]. RAI is insufficient glucocorticoid activity relative to the severity of the patient’s illness. RAI is usually seen in critically ill patients with low levels of serum cortisol and/or subnormal responses to adrenal stimulation. The definition of RAI does not encompass the subset of patients with apparently normal adrenal responses who appear to benefit from glucocorticoid supplementation. Therefore, a better definition is that of CIRCI. CIRCI is defined as a circulating level of cortisol inadequate for the severity of the patient’s illness and comprises both suppression of the HPA axis and also tissue resistance to the actions of cortisol [7]. During critical illness, inflammatory cytokines activate the HPA axis, resulting in a rise in serum cortisol. Annane et al. [13] demonstrated clearly that extremes in cortisol levels in this context, either low or high, are associated with poor prognosis. In addition, it appeared that patients with a subnormal response to challenge with synthetic ACTH also had a worse prognosis. These two findings suggest that, in a subset of critically ill patients, cortisol levels are inadequate relative to the severity of the systemic disease, or the adrenal glands are unable to respond adequately to the correct level of stimulus resulting in RAI. RAI/CIRCI arises from combined effects of both suppression of the HPA axis and resistance of tissue to circulating cortisol levels. Dysregulation of the HPA axis is multifactorial in critical illness, and causes include decreased ACTH response to CRH, inhibition of cortisol synthesis, and adrenal hypoperfusion. Although incompletely understood, the pro-inflammatory cytokine milieu has a central role in the process. Elevated levels of the pro-inflammatory cytokines tumor necrosis factor α (TNF-α) and interkeukin 1 (IL-1) have been shown to cause decreased ACTH response to CRH, and high levels of TNF-α can cause a reduction in cortisol synthesis by direct inhibition of the action of ACTH on the adrenal gland [14,15]. Furthermore, it has been shown that patients who demonstrate abnormal responses to exogenous ACTH also have higher levels of TNF-α [16]. Tissue resistance to glucocorticoids is thought to be due to downregulation of glucocorticoid receptor function and decreased transmission of activated receptors due to chronic exposure to pro-inflammatory cytokines such as TNF-α, IL-6, lipopolysaccharide (LPS), and macrophage migratory inhibitory factor [17–19]. This results in a picture consistent with adrenal failure, although levels of cortisol may be normal or even supranormal. Although widely documented in the literature, there is significant variation in reported incidence of RAI/CIRCI. This can be attributed to inconsistencies within patient populations studied, differences in diagnostic methods used to assess cortisol levels (serum total cortisol, serum free cortisol or other, including salivary cortisol levels), and a lack of concrete diagnostic criteria. Standardized methods of diagnosis have not been designed, and there is as yet no consensus regarding the optimal diagnostic tests or criteria required to determine the presence of RAI. Clinically, patients with cirrhosis have a hyperdynamic circulation, associated with decreased vascular resistance, hypotension, and increased cardiac output – a similar picture to that seen in patients with septic shock. Even in stable conditions, patients with cirrhosis demonstrate raised circulating levels of pro-inflammatory cytokines (TNF-α, IL-1, and IL-6) and endotoxin, which can have dysregulating effects on the HPA. Additionally, normal circadian rhythms within this population are disrupted by the effects of ammonia, which could affect the ability of the HPA axis to respond appropriately to stress [20,21]. Substrate depletion is an important contributor to adrenal dysfunction in liver disease. At times of stress most cortisol is synthesized from HDL cholesterol in the Apo-1 form, which is synthesized in the liver. The adrenal gland is therefore reliant on adequate substrate in order to produce levels of cortisol appropriate to the stress situation. Patients with cirrhosis have low levels of HDL cholesterol compared to controls – a finding that is proportional to disease severity as measured by Child–Pugh score [12]. The impact of this finding is observed in critically ill liver patients where adrenal responses correlate with baseline HDL levels and adrenal dysfunction can be seen to develop in patients with previously normal adrenal function and low HDL levels during prolonged critical illness – the adrenal exhaustion syndrome [10,22]. Another possible cause of AI in patients with cirrhosis is adrenal hemorrhage secondary to coagulopathy. In the study by Harry et al. [23], out of 12 patients that died, 8 had a postmortem and 1 patient was shown to have sustained adrenal hemorrhage (Figure 21.1). Figure 21.1 Pathophysiology of adrenal dysfunction in liver disease – The normal events leading to activation of the HPA and consequences of uncontrolled elevated levels of proinflammatory cytokines on the left of the figure. Factors that may be more relevant in patients with liver disease outlined on the right of the figure. ACTH, adrenocorticotropic hormone; CRH, corticotropin-releasing hormone; IL, interleukin; LPS, lipopolysaccharide; TNF, tumor necrosis factor. The true prevalence of RAI/CIRCI in cirrhosis is controversial as varying definitions have been used and there are inherent difficulties in measurement of cortisol in patients with liver disease. What is clear is that whichever definition is used the prevalence of the syndrome appears to correlate with disease severity and also carries prognostic significance. In patients with acute liver injury and liver failure, Harry et al. [23] found that the prevalence of AI was increased in line with the severity of liver disease and the degree of organ failure. In this study of 45 patients with either acute liver injury or acute liver failure, patients underwent adrenal function testing with the short Synacthen test (SST). An impaired response to adrenal stimulation was seen in 37% of patients with subnormal responses to adrenal stimulation seen more frequently in patients with more severe organ failure, need for emergency transplantation, and nonsurvivors. In patients with cirrhosis and septic shock. Tsai et al. [8] studied 101 septic cirrhotic patients, who underwent adrenal function testing. The results demonstrated that 51% of patients fulfilled their criteria for AI (baseline cortisol <9 μg/dL or an increment of <9 μg/dL after stimulation with 250 μg Synacthen). Patients with AI had lower mean arterial pressure and were more likely to need vasopressor support. Of note, a correlation was demonstrated between the degree of adrenal dysfunction and severity of disease, as per Child–Pugh, MELD, and Apache III scores. The response to adrenal stimulation was also an independent predictor of mortality, which was increased in the AI group. Using an alternative definition of AI, Marik et al. [10] reported the prevalence of AI within 340 patients admitted to a liver intensive care unit. The incidence of AI was found to be 66% in critically ill cirrhotic patients, with a prevalence of 33% in acute liver failure patients. In this study, the mortality rate of patients with AI was 39% overall. Patients who were subsequently treated with steroids had a mortality rate of 26% compared with 46% in those not treated with steroids, although this was not assessed in a randomized fashion. The use of hydrocortisone in cirrhotic patients with septic shock has now been assessed in a retrospective cohort study and a randomized controlled clinical trial [24,25]. In the first study, the effect of treating patients with cirrhosis and septic shock with low dose hydrocortisone was evaluated in a prospective fashion. Critically ill cirrhotic patients (n = 25) with either an inappropriately low random cortisol (<15 g/dL) or a subnormal (<9 g/dL) response to SST received 50 mg hydrocortisone four times daily. The patients treated with steroids were compared with a group of patients treated on the same intensive treatment unit prior to the introduction of adrenal function testing in septic shock. AI was diagnosed in 18/25 (68%). In the treated group of patients there was quicker resolution of shock and an increase in hospital survival compared to the 50 historical controls that had not undergone adrenal function testing (64% vs. 32%). In a randomized trial of patients with cirrhosis and septic shock, 75 patients underwent adrenal function testing and were randomized to receive intravenous 50 mg hydrocortisone four times daily or placebo within 48 hours of shock onset. Hydrocortisone therapy was continued until shock reversal (cessation of vasopressors) and the primary outcome was all cause 28-day mortality. The overall incidence of AI in the study population was 76% and was not significantly different between treated patients and controls. There was no difference in survival between the two groups although hydrocortisone-treated patients had a faster resolution of shock. However, in hydrocortisone-treated patients there was a higher incidence of shock relapse and gastrointestinal bleeding. The incidence of shock relapse in this study was much higher than in other studies using low dose hydrocortisone suggesting that one factor leading to the overall negative result may have been the abrupt cessation of steroid supplementation rather than tapering. This could be important in patients with cirrhosis if one considers that AI may be a pre-existing condition rather than a result of the sepsis per se.
Adrenal Insufficiency
Introduction
Background
Physiology of the Hypothalamic–Pituitary–Adrenal Axis
Relative Adrenal Insufficiency and Critical Illness-Related Corticosteroid Insufficiency
Pathophysiology
Adrenal Insufficiency and Liver Disease
Prevalence and Clinical Relevance of Adrenal Insufficiency in Liver Disease
RAI/CIRCI in Critically Ill Patients with Liver Disease
Prevalence of Adrenal Insufficiency in Stable Cirrhosis
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


