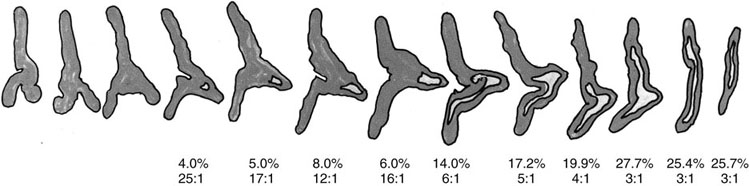
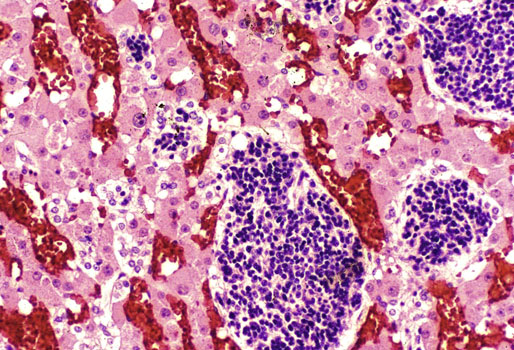
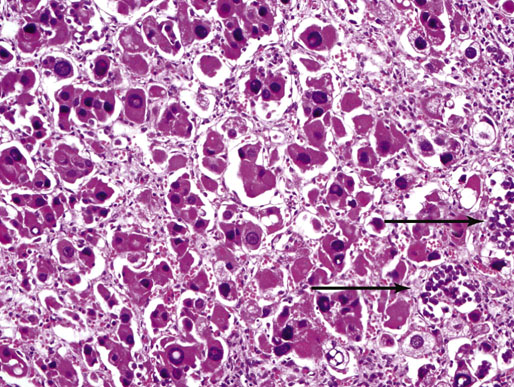
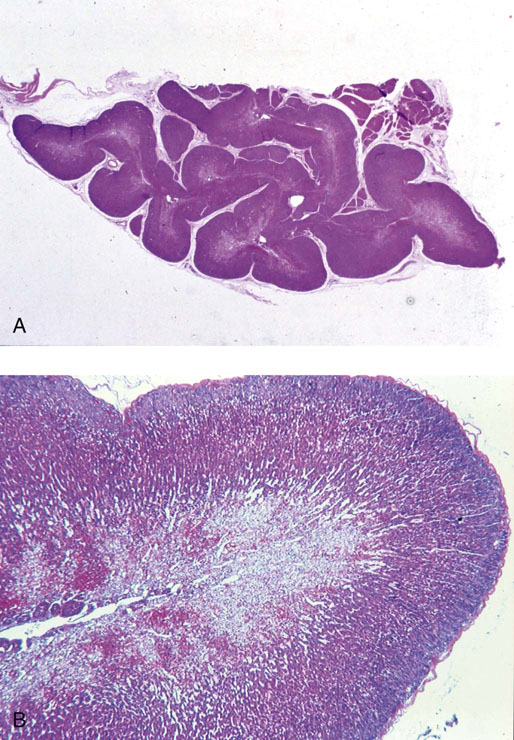
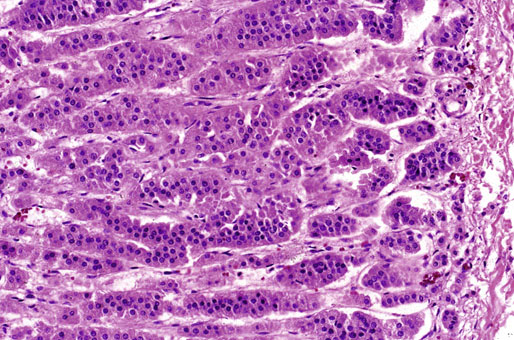
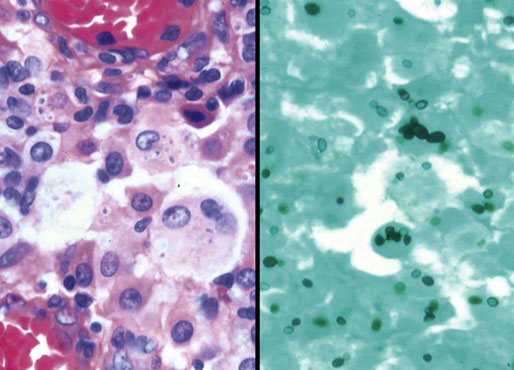
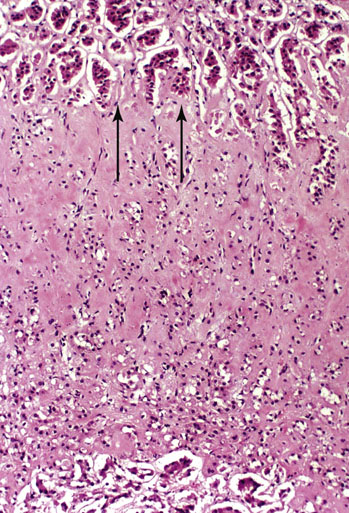
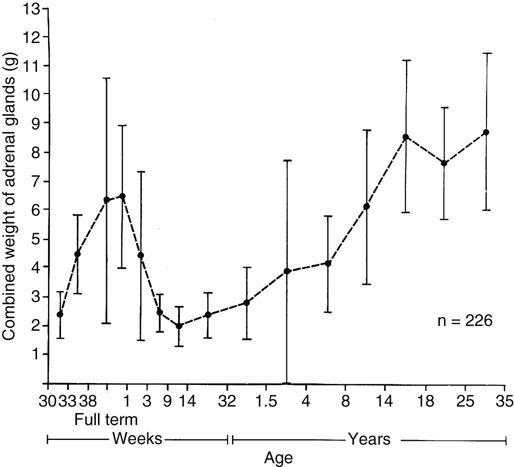
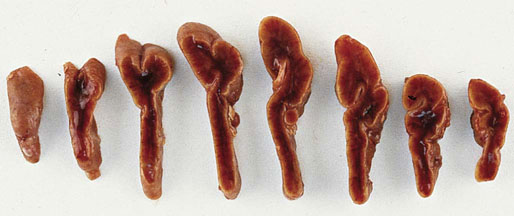
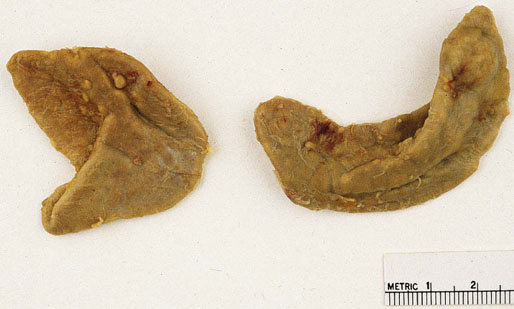
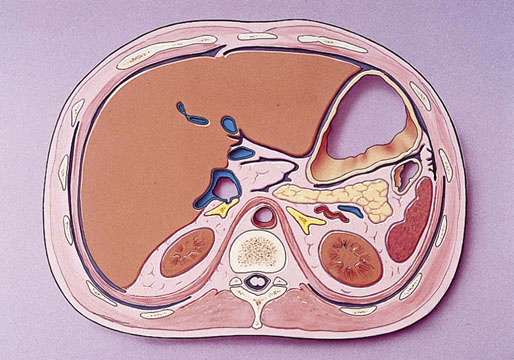
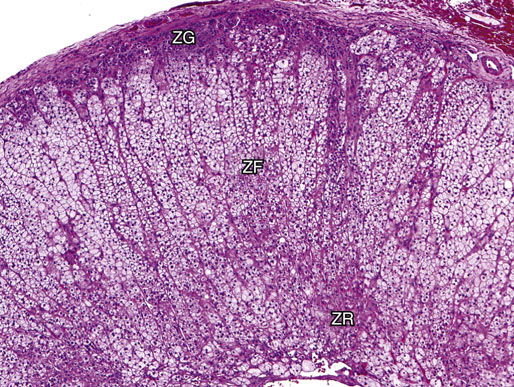
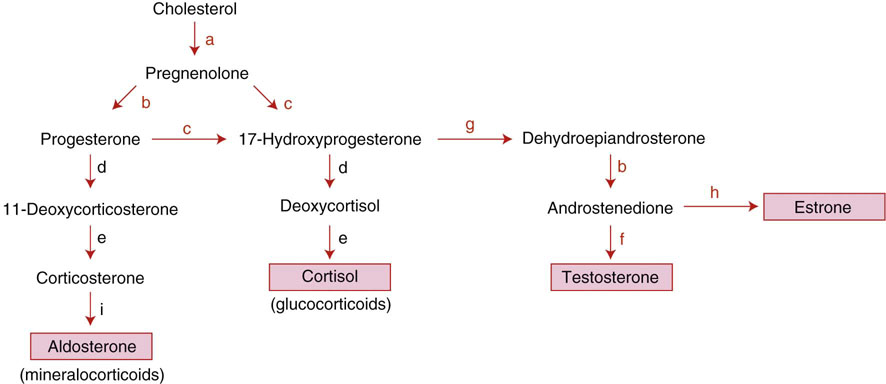
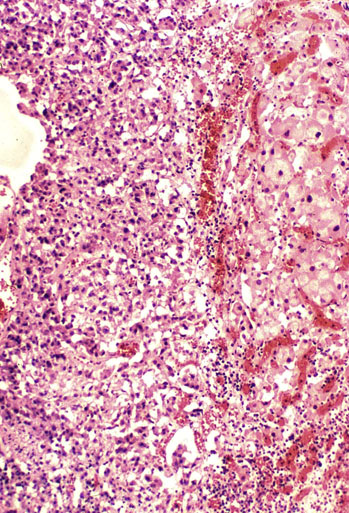
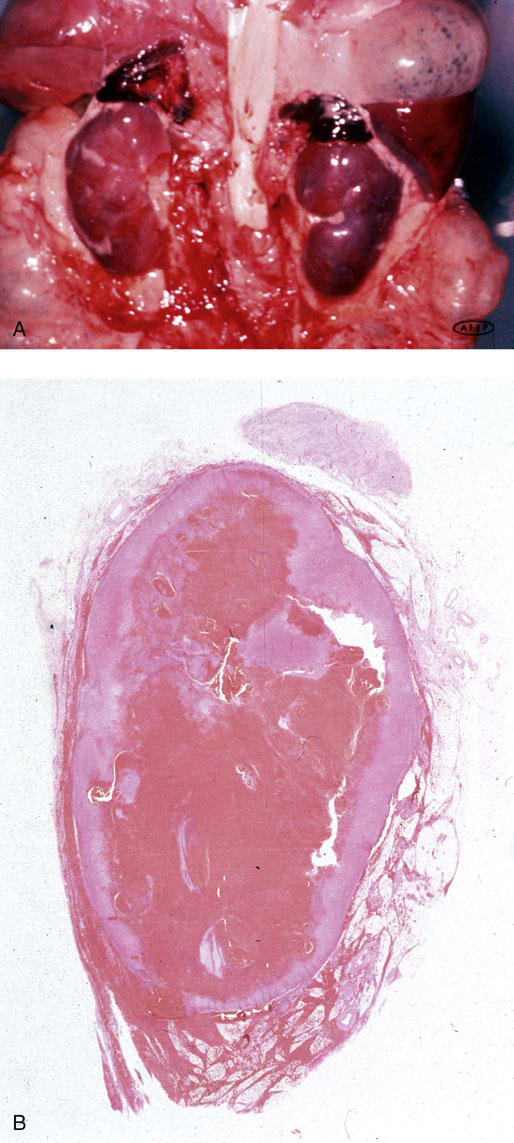
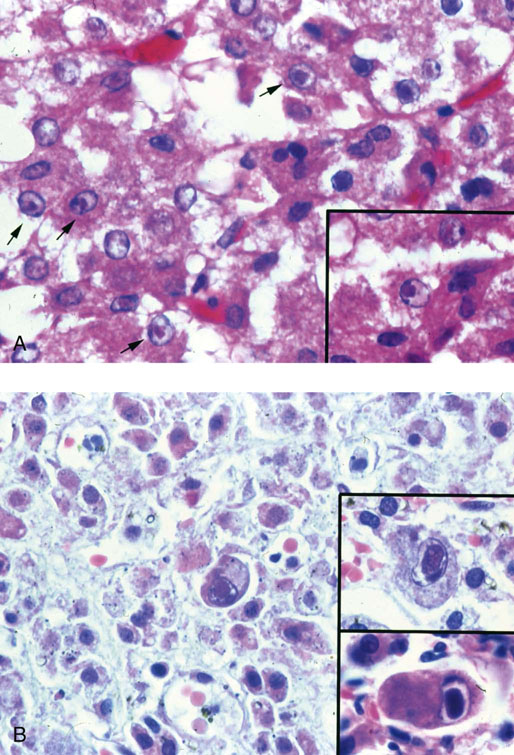
Chapter 16 Embryology and normal gross anatomy Congenital and other abnormalities Adrenal aplasia and hypoplasia Congenital adrenal hyperplasia Stress-related changes of the adrenal gland Chronic adrenal cortical insufficiency (Addison disease) Acute adrenal cortical insufficiency Inflammation and other infections Multiple endocrine neoplasia type 1 Other rare causes of Cushing syndrome Adrenal cortical hyperplasia with hyperaldosteronism Adrenal cortical hyperplasia with excess sex steroid secretion Neuroblastic tumors: neuroblastoma, ganglioneuroblastoma, and ganglioneuroma The primordium of the adrenal cortex becomes evident at Carnegie stage 14 (~5 to 7 mm and 32 days), just lateral to the base of the dorsal mesentery near the cranial end of the mesonephros.1,2 The adrenal cortical primordia are of mesodermal origin, and during development in the late embryo and fetus the portion of the developing cortex that occupies the greatest volume is referred to as the fetal or provisional cortex. This layer of cortex makes up approximately 80% of the newborn adrenal gland and undergoes marked regression in the first weeks of life; this is shown graphically in Figure 16-1, in which the combined weight of the adrenal glands decreases by almost 50% by the ninth to the fourteenth week after birth.3 The adult, or definitive, cortex forms a much thinner outer zone beneath the adrenal capsule and ultimately becomes the trilayered adrenal cortex of the adult. Convincing evidence exists for centripetal migration or displacement of adrenal cortical cells in experimental animals, thus supporting the original cell migration theory of Gottschau that proposed that migrating adrenal cortical cells were capable of producing all of the major adrenal cortical steroids.3 Fig. 16-1 Average combined weight of the adrenal glands from 226 autopsies by age. Note the marked reduction in combined weight in the first few weeks of life caused by regression of the fetal (provisional) cortex. (From Lack EE, Kozakewich HPW. Embryology, developmental anatomy, and selected aspects of nonneoplastic pathology. In: Lack EE, ed. Pathology of the adrenal glands. New York: Churchill Livingstone, 1990;1-74.) On gross examination the late fetal or neonatal adrenal gland is relatively soft, and in transverse sections the fetal zone may have rather dark coloration, which at this stage is quite broad. This dark appearance, shown in Figure 16-2, may be misinterpreted as adrenal hemorrhage or apoplexy. The adrenal glands in newborns have smoother external surfaces than in adults. In the adult, the right adrenal gland is roughly pyramidal and the left is more elongated (Fig. 16-3). Inspection of the intact capsular surface of the gland after removal of periadrenal connective tissue and fat may reveal small capsular extrusions of cortex; some of these are directly connected with the underlying cortex, but others seem to lie free on the capsular surface or unattached in periadrenal fat. Transverse sections of adrenal gland in the adult reveal a bright yellow, relatively uniform cortex with a gray-white medulla that is concentrated in the head and body of the gland (Fig. 16-4). A cuff of cortical cells may be noted partially or entirely surrounding larger tributaries of the adrenal vein. The dorsal surface of the adrenal gland has a longitudinal ridge or crista flanked by medial and lateral extensions or alae (wings). The anterior (or ventral) surface of the adrenal gland is relatively smooth, and it is from this surface of the gland that the adrenal veins exit and drain into the inferior vena cava on the right side and the renal vein on the left. The orientation of the adrenal gland in vivo differs from that depicted in gross photographs of specimens in surgical or autopsy material. As seen in Figure 16-5, the glands are oriented in a more vertical axis, with the ridge (or crista) projecting posteriorly and flanked by medial and lateral alae. The thickness of the adult cortex is approximately 2 mm or more throughout most of the gland, although some variability may be seen from area to area, and cortical nodularity may complicate the morphology.3 Fig. 16-2 Adrenal gland from a newborn infant. Note the dark congested fetal (provisional) cortex and thin rim of pale adult (definitive) cortex. Cortical extrusion is also present centrally. Fig. 16-3 Normal adrenal glands from an adult. The right is roughly pyramidal (left side of photo), whereas the left is elongated (right side of photo). The longitudinal ridge (crista) is flanked by lateral extensions (alae). Fig. 16-4 Normal adrenal from an adult. An incomplete cuff of cortical cells is present around a central adrenal vein in the medulla. The adrenal vein is present on the ventral surface toward the head of the gland. The dorsal ridge (crista) is flanked by lateral and medial extensions (alae). Medullary tissue is concentrated in the body and head and appears gray-white, in contrast to the bright yellow cortex. Fig. 16-5 Diagram of transverse cut of abdomen in an adult showing orientation of the adrenal glands and relation with kidneys. The ventral aspect of both glands is relatively flat, and dorsal surface has a longitudinal ridge (crista). Lateral extensions (alae) are often referred to as medial and lateral limbs on CT scans. Chromaffin tissue of the adrenal medulla in the fetal and newborn adrenal gland is inconspicuous on gross examination of transverse sections of the gland. In the adult, however, chromaffin tissue is concentrated in the body and head of the gland, with the latter regions being directed inferomedially in vivo.3,4 As seen in Figure 16-6, the ratio of area occupied by cortex relative to that of medulla decreases considerably from the tail to the body and head of the gland. The normal overall ratio of cortex to medulla is approximately 10 : 1. The distribution and amount of chromaffin tissue within the gland, as well as other factors, such as adrenal weight, may be important in determining whether adrenal medullary hyperplasia is present.4 Another consideration is morphologic abnormalities of the adrenal cortex, such as adrenal cortical atrophy, which can affect the overall ratio. The adrenal medulla is composed of chromaffin cells derived from primitive sympathicoblasts of neural crest origin that migrate into the dorsomedial aspect of the adrenal primordium and become apparent at Carnegie stages 16 and 17 (~11 to 14 mm and 41 days).1,2 Most of the developing chromaffin tissue in fetal life is extraadrenal, with the largest collections of cells in the paraaortic region, near the origin of the superior mesenteric and renal arteries down to the aortic bifurcation; these chromaffin structures were first characterized in the human fetus by Zuckerkandl5 in 1901, who referred to them as the aortic bodies. At birth, the thin rim of adult (definitive) cortex blends imperceptibly into cells of the fetal (provisional) cortex. The definitive cortex apparently begins to grow soon after birth,6 with zonation into the zona glomerulosa and zona fasciculata first appearing at 2 to 4 weeks. According to some investigators, the zona reticularis appears at approximately 4 years,6 but others contend that it appears before 1 year.7 The zona glomerulosa contains cells with dark round nuclei and relatively scant cytoplasm arranged in interlacing cords and spherules; this zone is normally thin and ill defined, and it may appear discontinuous in the normal adult gland (Fig. 16-7). This layer blends imperceptibly into the zona fasciculata, which constitutes most of the adult cortex, and consists of long columns of larger cells with pale, finely vacuolated cytoplasm in the unstressed gland. The transition between the innermost zona fasciculata and the zona reticularis contains cells with more compact eosinophilic cytoplasm separated by thin-walled sinusoids and irregular short cords of cells. Reticularis cells may contain prominent granular pigment representing lipofuscin. Chromaffin cells of the adrenal medulla are polyhedral and arranged in short anastomosing cords or nests with a prominent vascular network, or a more solid or diffuse arrangement of cells may be seen. Adrenal chromaffin cells secrete predominantly epinephrine, with lesser amounts of norepinephrine.4 In the fetal and neonatal adrenal, small nests of primitive neuroblastic cells may be encountered (Fig. 16-8), which may be a part of normal developmental anatomy3,4 (see later discussion of in situ neuroblastoma). Fig. 16-7 Normal adult adrenal cortex. Zona glomerulosa (ZG) is at the top of the field beneath the adrenal capsule and forms a thin, discontinuous layer of cells. Most of cortex is occupied by radial interconnecting cords of zona fasciculata (ZF), which contain cells with pale-staining, lipid-rich cytoplasm. The zona reticularis (ZR) has interconnecting short cords of cells with compact, eosinophilic cytoplasm and congested microvasculature. The zona glomerulosa is the site of aldosterone production and is responsive to stimulation by angiotensin and adrenocorticotropic hormone (ACTH). The zona fasciculata produces corticosteroids such as cortisol, whereas the zona reticularis is responsible for sex steroid production. Longitudinal pillars of smooth muscle are found predominantly in the head of the adrenal gland around tributaries of the adrenal vein and are thought to act as “sluice gates” that retard the flow of blood from the medullary venous sinuses and plexus reticularis during muscle contraction.8 The muscular bundles may help regulate medullary blood flow and influence the degree of congestion in the zona reticularis and zona fasciculata of the adjacent cortex. Examination of the intact adrenal gland is best accomplished by careful removal of as much investing connective tissue and fat as possible to obtain an accurate weight. The weight of the cleanly dissected gland may provide valuable information regarding adrenal cortical or medullary pathology. In the study by Stoner and colleagues,6 the average combined weight of the adrenal glands at birth was 10 g (range, 2 to 17 g), whereas the average weight was 6 g at 7 days of age and 5 g at 2 weeks of age. Quinan and Berger9 studied the adult adrenal, concentrating on ostensibly healthy subjects who had died suddenly, and found that the average weight of each gland was 4.15 g, with no significant difference between the right and left sides. Studzinski and associates10 examined surgically removed adrenal glands from women with breast cancer and reported an average weight of 4 g, with little variation (standard deviation, 0.8 g). Adrenal glands obtained at autopsy from individuals who had not died suddenly or unexpectedly tended to be heavier, with an average individual weight of 6 g; this difference was attributed to the stress of illness and the trophic influence of endogenous ACTH.10 Using these data, each normal adrenal gland should weigh less than 6 g, provided excess periadrenal fat and connective tissue are carefully removed. Complete bilateral adrenal aplasia is rare and may occur in a familial setting.11 The four forms of congenital adrenal hypoplasia are (1) a sporadic form associated with hypopituitarism, (2) an autosomal recessive form, (3) an X-linked cytomegalic form associated with hypogonadotropic hypogonadism, and (4) a form associated with glycerol kinase deficiency, a condition that is also X linked.12–14 The hereditary form is divided into a miniature type, which affects both sexes and causes extremely small adrenal glands, with normal architecture, and the more common cytomegalic type, which has an X-linked pattern of inheritance.3 In the cytomegalic type, affected males have small adrenal glands that are architecturally abnormal and contain scattered cytomegalic cells that are sometimes vacuolated and may contain intranuclear pseudoinclusions. The onset of adrenal cortical insufficiency is variable and depends chiefly on the endocrine reserve of the existing adrenal cortex. Affected infants may present in the newborn period with weight loss, vomiting, dehydration, and a tendency toward severe salt loss. Prompt recognition of this rare adrenoprival condition results in improved survival. Males with the X-linked cytomegalic form of adrenal hypoplasia typically have hypogonadotropic hypogonadism in adolescence. A recently discovered mutation in the NR0B1 gene encoding the DAX1 protein gives rise to X-linked adrenal hypoplasia congenita and is also believed to play a role in hypogonadotropic hypogonadism.14–19 Adrenal insufficiency has been reported in several other conditions, including familial glucocorticoid deficiency, glycerol kinase deficiency, and selective hypoaldosteronism.3 During embryologic development the adrenal primordium is in close proximity to the urogenital ridge, accounting for the accessory and heterotopic adrenal tissue that may occur in sites in the upper abdomen and along lines of descent of the gonads.3 Heterotopic adrenal tissue has been described in up to 32% of patients in the region of the celiac axis, and at this site approximately half of the lesions contained both cortex and medulla.20 Accessory adrenal tissue in sites further removed from the upper abdomen usually consists of cortical tissue alone, without the distinctive zonation of the normal adult adrenal gland. Other sites of heterotopia include the broad ligament near the ovary (23%),21 the liver,22 the kidney (6%, usually subcapsular),23 along the spermatic cord (3.8% to 9.3%; a higher incidence was observed for males undergoing surgery for an undescended testis),24,25 the testicular adnexa (7.5%),26 and other rarely described sites that defy ready embryologic explanation, such as the placenta,27 lung,28,29 mediastinum,30 and an intracranial intradural location.31 Only rarely have intratesticular32 or intraovarian33 cortical rests occurred within the substance of the gonads. Cases have been reported of hyperplastic cortical nodules arising from accessory adrenal tissue along the spermatic cord and the broad ligament,34 and adrenal cortical neoplasms have been reported, rarely, in hepatic parenchyma35 and the spinal canal.36–38 Intrarenal adrenal heterotopia could potentially be confused with low-grade clear cell renal cell carcinoma. 23 Union or adhesion of the adrenal gland to kidney or liver has been reported, the distinction being whether a continuous connective tissue capsule separates the two organs.39 Adrenal fusion is a rare anomaly in which the adrenal glands are fused in the midline and may be associated with other congenital midline defects such as spinal dysraphism or indeterminate visceral situs.3 Abnormal adrenal shape has been reported in some cases of renal agenesis, in which the glands may be ovoid with smoother contours.3 Congenital adrenal cytomegaly is usually an incidental finding in an adrenal gland that otherwise appears grossly normal. It has been reported in approximately 3% of newborn autopsies and 6.5% of premature stillborns.40 The cytomegaly affects cells of the fetal (provisional) cortex and may be bilateral or unilateral, focal or diffuse. The cytomegalic cell has an enlarged hyperchromatic nucleus and increased volume of cytoplasm. Nuclei may be markedly pleomorphic and occasionally contain intranuclear pseudoinclusions, which are indentations of the nucleus with invagination of cytoplasm. Despite the marked nuclear abnormalities, mitotic figures are characteristically absent. Adrenal cytomegaly is a characteristic component of the Beckwith-Wiedemann syndrome (Fig. 16-9),41–43 sometimes called the EMG syndrome, which refers to a major triad of findings—exomphalos, macroglossia, and gigantism.3 The estimated frequency of this disorder is 1 in 13,000 births; most reported cases are sporadic, although some seem to have a mendelian pattern of inheritance.3 The disorder is caused by dysregulation of growth regulatory genes within the 11p15 region, resulting in loss of normal growth control and increased incidence of certain cancers.44,45 The adrenal glands in this disorder are enlarged, with combined weights as high as 16 g. The adrenal cytomegaly is usually marked and is typically bilateral and diffuse. Curiously, adrenal chromaffin tissue may be hyperplastic or inappropriately mature.4,43 Visceromegaly may be present, affecting the kidneys and pancreas, and some infants develop severe neonatal hypoglycemia that may prove fatal. An increased incidence of malignant tumors is seen in this disorder, usually Wilms tumor or adrenal cortical carcinoma, but other neoplasms have been reported, including neuroblastoma, pancreatoblastoma, and hepatoblastoma.3,46–49 The presence of hemihypertrophy in children predicts a greater risk for the development of a malignant neoplasm. Adrenal cytomegaly has been associated with fetal hydrops resulting from Rh incompatibility or hemoglobin Bart.50 Adrenoleukodystrophy occurs in multiple forms. Most cases of adrenoleukodystrophy are X linked. The molecular basis of X-linked adrenoleukodystrophy is mutation of the gene (ABCD1) encoding a peroxisomal membrane protein (ALDP), resulting in an increase in very-long-chain fatty acids (VLCFA) and reduced oxidation thereof in the peroxisomes.51–54 The lipid material accumulates in tissue as cholesterol esters that may exert a toxic effect on cells, with crystallization of lamellae and disruption of cell membranes.3 This mechanism may account for the pathogenesis of adrenal cortical insufficiency and degeneration of white matter, particularly involving the posterior cerebrum, cerebellum, and descending corticospinal tracts. Three main forms of X-linked adrenoleukodystrophy are known: (1) the childhood form, which is a progressive central demyelinization leading to total disability within 2 years of onset; (2) the adult form, also known as adrenomyeloneuropathy, in which a slowly progressive spastic paraparesis and distal polyneuropathy occur, with onset usually in the second or third decade of life3; and (3) a variant that has a primary presentation of adrenal insufficiency. Adrenoleukodystrophy or adrenomyeloneuropathy should be considered in the differential diagnosis of boys or young men who present with unexplained adrenal cortical insufficiency, because neurologic symptoms may not be evident at the time of presentation.51–54 A rare fourth neonatal form usually has an autosomal recessive inheritance.3 The adrenal glands in adrenoleukodystrophy are often quite small. Thinning of the adrenal cortex occurs, with characteristic enlargement of cortical cells having abundant ground-glass or waxy cytoplasm55; these cells have been called balloon cells and may have a fibrillar or striated appearance caused by lipid material being extracted during routine processing. On ultrastructural examination, bilamellar and lamellar lipid inclusions can be seen, which are virtually pathognomonic for the disorder. The adrenal cortical insufficiency in this disorder is primary and not caused by pituitary or hypothalamic dysfunction. The recent molecular advances regarding the X-linked form of this disease have major implications for the possibility of gene-based therapy.51,54,56 Congenital adrenal hyperplasia (adrenogenital syndrome) is a rare autosomal recessive disorder caused by a deficiency of one of five different enzymes in the steroid biosynthetic pathway of the adrenal cortex (Fig. 16-10).3,57 The first unmistakable and thorough description of congenital adrenal hyperplasia was in 1865 by the Italian anatomist de Crecchio,58 who dissected the cadaver of an apparent male of approximately 40 years of age who had bilateral cryptorchidism and partial hypospadias; however, further examination revealed a vagina, uterus, fallopian tubes, ovaries, and very large adrenal glands. The subject was also said to have had frequent diarrhea and vomiting and in his final days had extreme weakness and exhaustion, which almost certainly represented an addisonian crisis. Fig. 16-10 Normal biosynthetic pathway of adrenal cortical steroid synthesis. a = 20,22 hydroxylase and 20,22 desmolase; b = 3β-hydroxysteroid dehydrogenase; c = 17-hydroxylase; d = 21-hydroxylase; e = 11β-hydroxylase; f = 17-ketosteroid reductase; g = 17,20 desmolase; h = P450 aromatase; i = 18-hydroxylase and 18-aldehyde synthetase. The most common cause of congenital adrenal hyperplasia is 21-hydroxylase deficiency, which accounts for approximately 90% to 95% of cases.59,60 This deficiency has been divided into a classic form, with an incidence of 1 in 5000 to 1 in 15,000 live births in most white populations; a nonclassic form, which is among the most frequent of autosomal recessive disorders in the white population; and a cryptic form in which patients are asymptomatic despite having the same biochemical abnormality.59 Mutations in the encoding gene have been confirmed as the basis of endocrine disease in all adrenal steroidogenic enzymes required for the synthesis of cortisol, except for cholesterol desmolase.60 In 21-hydroxylase deficiency, insufficient production of cortisol occurs, resulting in a lack of negative feedback at the hypothalamic–pituitary level and secondary trophic stimulation of the adrenal glands by increased ACTH levels. In approximately two thirds of cases of the classic disorder, biosynthesis of aldosterone is also affected, leading to “salt wasting”; if unrecognized or severe enough, this may result in death in the first few weeks of life.3 The remaining third of cases have simple virilizing disease, without significant impairment of aldosterone biosynthesis. Because of the enzymatic block, precursor steroids accumulate and are diverted into the sex steroid pathway with increased androgen production. The development of external genitalia is under the control of androgens in utero, and, because of this, affected females usually have ambiguous genitalia with clitoromegaly and fusion of the labioscrotal folds, although the internal female organs develop normally. In females the ambiguous genitalia may be so extreme that the child is incorrectly assumed to be male. Affected males usually appear normal at birth. For this reason, males with the disorder may not be diagnosed until they present with more severe symptoms, often related to a potentially fatal salt-wasting crisis. The identification of mutations in the CYP21 gene, which encodes the 21-hydroxylase enzyme, has led to advances in DNA diagnosis, including prenatal and newborn screening programs.61–63 Approximately 5% to 8% of cases of congenital adrenal hyperplasia are caused by the classic form of 11β-hydroxylase deficiency63 as a result of mutations of the CYP11B1 gene.64,65 Other enzymatic defects causing congenital adrenal hyperplasia include deficiency of 3β-hydroxysteroid dehydrogenase, 17α-hydroxylase, and the rare deficiency of cholesterol desmolase, the enzyme involved in early steroidogenesis. Today it is very uncommon to examine the adrenal glands of patients who died of unrecognized or untreated congenital adrenal hyperplasia. Grossly, the adrenal glands are enlarged, often with a convoluted or cerebriform surface with excess cortical plications and folding. The glands frequently have a light brown color reminiscent of the zona reticularis, resulting from the sustained trophic effect of ACTH and the conversion of lipid-rich, pale-staining cells to cortical cells, which have lipid-depleted compact eosinophilic cytoplasm (Fig. 16-11). In children, the weight of each adrenal gland may be 10 to 15 g, whereas in older individuals each gland may weigh 30 to 35 g.3 Cholesterol desmolase (cytochrome P450scc [side chain cleavage]) deficiency is also called congenital lipoid hyperplasia because accumulation of cholesterol and cholesterol esters occurs, which gives the gland a bright-yellow or white appearance on cross section. Male patients with congenital adrenal hyperplasia—particularly but not exclusively the salt-losing form of 21-hydroxylase deficiency—may develop one or more testicular tumors. By adulthood, up to 94% of patients develop these tumors, but occasionally they present earlier, in adolescence or childhood.66–68 The tumors are often bilateral (83%) and may cause testicular pain and tenderness.69 Several reports in the literature clearly document that the tumors are ACTH dependent, as evidenced by reduction in testicular size and associated symptoms with suppressive doses of dexamethasone and recrudescence of testicular enlargement with ACTH stimulation. Laboratory testing also has demonstrated ACTH-dependent steroidogenesis by the tumors.70 The tumors may be 2 to 10 cm in diameter in older patients. Most of the smaller tumors appear to be located in the hilum of the testis, but with larger tumors the precise site of origin is difficult to determine.69 On cross section the tumors often have a lobulated appearance with bulging, tan to dark brown nodules. The histologic appearance resembles that of a Leydig cell tumor; however, some features may distinguish these two tumors. Nuclei are usually round to oval with a single prominent nucleolus, which may be central or somewhat eccentric. Cells have granular, pink cytoplasm with relatively distinct cell borders. The cells are usually arranged in sheets or small nests with intersecting fibrous bands, and reticulum stain often demonstrates an intimate pattern of isolation of individual and small clusters of cells. As opposed to Leydig cell tumors, testicular adrenogenital tumors lack Reinke crystalloid, exhibit more extensive fibrosis, and frequently contain lymphoid aggregates. Nuclear pleomorphism is reportedly prominent in contrast to Leydig cell tumors, and mitotic activity is rare. Most testicular adrenogenital tumors are strongly positive for synaptophysin, whereas only rare Leydig cell tumors are immunoreactive with synaptophysin.71 Virtually identical testicular tumors of this type have been reported in males with Nelson syndrome (pituitary adenoma after bilateral adrenalectomy)72; rarely, a female with Nelson syndrome may develop similar adrenal rest tumors in the region of the ovaries where heterotopic adrenal cortical tissue occasionally is found.34 The histogenesis of these interesting testicular tumors is not clear; possibilities include origin from Leydig cells, adrenal cortical rests, or multipotent testicular stromal cells capable of differentiation into either Leydig or adrenal cortical cells, depending on the hormonal milieu. It is unclear whether these tumors are true neoplasms or hyperplastic nodules. In favor of hyperplasia are their ACTH dependence and bilaterality. Rare cases of a histologically similar tumor arising in the ovary of women with congenital adrenal hyperplasia resulting from 21-hydroxylase deficiency have been reported.73,74 Rare cases of adrenal cortical adenoma and carcinoma have been reported in patients with congenital adrenal hyperplasia.3,75,76 It has been suggested that persistent ACTH stimulation may result in neoplastic transformation of some adrenal cortical cells, but this is unproved. Other tumors have been reported in association with congenital adrenal hyperplasia, including bilateral adrenal myelolipomas,77 osteosarcoma, and Ewing sarcoma,78 but their relationship with congenital adrenal hyperplasia and the underlying biochemical abnormality is unclear. One of the most common histologic changes observed in the adrenal gland of patients under stress is the conversion of lipid-rich, pale-staining cortical cells of the zona fasciculata to cells with compact, lipid-depleted eosinophilic cytoplasm. This is particularly common in acquired immunodeficiency syndrome (AIDS) (Fig. 16-12). Another abnormality reported in stress-related conditions is degeneration of the outer zona fasciculata, initially described as tubular degeneration; this abnormality appears with scattered necrosis of cortical cells, shedding of vacuolated cytoplasm, and exudation of fluid into cords of cortical cells in the outer zona fasciculata.79 A peculiar vacuolization of the fetal adrenal cortex has been described in infants with erythroblastosis fetalis, and nearly identical changes have been observed in thalassemia major.3 A relationship with intrauterine stress and hypoxia has been suggested. A pattern of focal lipid depletion has been reported as lipid reversion, which suggests recovery from stress and replenishment of lipid in cells of the inner zona fasciculata. In areas of lipid reversion the outer aspect of the adrenal cortex contains little or no lipid, although lipid is prominent in the inner zona fasciculata.7 Conspicuous iron accumulation occurs in the form of hemosiderin in cells of the outer cortex, particularly the zona glomerulosa, in conditions such as primary hemochromatosis and transfusion hemosiderosis.3 In some cases, associated hypothalamic–pituitary dysfunction is caused by excess iron deposition, which may result in endocrine insufficiency of the gonads, thyroid, and adrenal glands. A variety of drugs and cytotoxic agents have direct antiadrenal activity; examples include dichlorodiphenyltrichloroethane (DDT) and its derivative o,p′DDD, which has been used for palliative treatment of patients with adrenal cortical carcinoma because of its adrenolytic effect on normal and neoplastic cortical cells. Another agent with antiadrenal activity is ketoconazole, a broad-spectrum antifungal drug that blocks adrenal steroid synthesis.3 Linear hyaline fibrosis has also been reported in the zona reticularis after radiation, probably because of structural damage of the vascular plexus in this zone.80 Anencephaly is a severe developmental defect of anterior neural tube structures with agenesis of much of the brain and cranial vault. The pituitary gland is difficult to find grossly but is often identified in histologic sections, albeit reduced in amount. The adrenal glands are often extremely small in this disorder, with an average combined weight in one study of 1.8 g, but a significant number weighed less than 1 g.81 The fetal cortex is often normal in size and structure until approximately 20 weeks of gestation, after which it progressively involutes, similar to changes that normally occur after birth; chromaffin tissue may appear relatively prominent. The most common type of Addison disease is idiopathic or autoimmune and is regarded as an organ-specific autoimmune form of adrenalitis. It may be isolated or part of an autoimmune polyendocrinopathy syndrome. It has been suggested that Addison disease has a strong genetic component, including autoimmune polyendocrinopathy syndrome and, more often, expression of disease susceptibility, alleles associated with organ-specific autoimmunity (MHC, CTLA4, PTPN22), and those that encode proteins of innate immune response.82 It has been shown that presence of autoantibodies directed against the enzymes of steroid hormone production, predominantly 21-hydroxylase, and their detection may be helpful in predicting the risk for developing the disease.82–85 The adrenal gland in this disorder may be greatly reduced in size and volume, making gross identification difficult at autopsy unless numerous tissue blocks from the suprarenal bed are examined. The adrenal cortex has a large endocrine reserve, and it is estimated that up to 90% or more of the cortex must be ablated before functional impairment is apparent.3 In some cases, intercurrent illness, infection, or surgery may precipitate an addisonian crisis. The residual cortex is often thin and discontinuous, with scattered islands of cortical cells (Fig. 16-13) admixed with lymphocytes, plasma cells, and occasional lymphoid follicles, sometimes with reactive germinal centers. Cortical cells may be enlarged, with ample compact, eosinophilic cytoplasm and occasional nuclear alteration, including pseudoinclusions. On occasion, residual cortex may be difficult to identify, and little or no inflammation may be present. Fig. 16-13 Primary idiopathic or autoimmune form of Addison disease. Atrophic adrenal cortex (right side) contains cells with compact, eosinophilic cytoplasm. The medulla is present on the left. Autoimmune polyendocrinopathy syndrome (also known as autoimmune polyendocrinopathy–candidiasis–ectodermal dystrophy) is a rare autosomal recessive disorder with endocrine insufficiency involving various organs, including the adrenal cortex. Numerous mutations in the gene responsible for the disease, AIRE (autoimmune regulator), have been identified. The gene is located on chromosome 21 (21q22) and encodes a protein involved in transcriptional regulation.86–89 This disorder also has been called autoimmune polyglandular syndrome type I, and has a variety of manifestations.90 The combination of nontuberculous Addison disease and thyroid insufficiency is referred as Schmidt syndrome.91 Adrenal tuberculosis once was the leading cause of Addison disease.92 According to the review by Guttman93 of cases between 1900 and 1929, 70% resulted from tuberculosis, and 19% were caused by primary or idiopathic atrophy. The endemic nature of bovine tuberculosis in the early decades of the twentieth century is reflected in the comment by Dunlop,94 who characterized cream on top of the milk in those days as often being composed of “tuberculous pus.” The tuberculous adrenal is often enlarged, with extensive areas of caseous necrosis. A classic granulomatous reaction with numerous epithelioid histiocytes is present typically in extraadrenal sites but not the adrenal, suggesting that the locally high levels of adrenal corticosteroids dampen the host inflammatory response.3 Disseminated histoplasmosis is a recognized cause of Addison disease and typically involves the glands bilaterally. In a study of almost 100 cases, 7% of patients had chronic adrenal cortical insufficiency.95 Similar to tuberculosis, extensive caseous necrosis is a common finding, although a granulomatous response is the exception rather than the rule. Perivasculitis involving extracapsular adrenal vessels may lead to extensive infarction and caseous necrosis, resulting in loss of adrenal parenchyma and development of Addison disease. Other mycotic infections causing Addison disease are less frequent, including North American blastomycosis, South American blastomycosis, coccidioidomycosis, and paracoccidioidomycosis.3,96 On microscopic examination the organisms are often found clustered within the cytoplasm of macrophages (Fig. 16-14). They are spherical to oval and typically have single buds attached by a relatively narrow base. The adrenal may be involved in primary and secondary (AA) forms of amyloidosis. In primary amyloidosis, involvement of arterioles tends to occur, whereas in the secondary form there is usually extensive involvement of the cortex by the characteristic homogeneous eosinophilic material, resulting in severe atrophy and distortion of cells in the zona fasciculata and zona reticularis (Fig. 16-15). In advanced cases, cortical function can be impaired.97 Acute adrenal cortical insufficiency can occur in the setting of systemic infection with Waterhouse-Friderichsen syndrome but is seldom documented by laboratory or biochemical studies. Waterhouse-Friderichsen syndrome is seen classically in meningococcemia, but occasionally other bacteria, such as Streptococcus species, staphylococci, Rickettsiae, Ehrlichia, Clostridium species, Klebsiella species, Legionella species, Bacillus anthracis, and Treponema pallidum,98 are responsible. The course of the disease is usually fulminant, with a fatal outcome within 48 hours of onset, accompanied by mucocutaneous petechial hemorrhages and vascular collapse. The adrenal is usually intensely hemorrhagic, with confluent areas of coagulative necrosis, often associated with small fibrin deposits within sinusoids; occasionally more extensive hemorrhage occurs, with expansion of the gland and periadrenal hemorrhage (Fig. 16-16). Fig. 16-16 Adrenal glands in Waterhouse-Friderichsen syndrome. A, In situ photograph showing bilateral hemorrhagic adrenal glands in a suprarenal location (autopsy specimen, from files of Armed Forces Institute of Pathology, Washington, DC). B, Extensive necrosis and hemorrhage are seen in this adrenal gland and extend into periadrenal connective tissue. (From Lack EE, Kozakewich HPW. Pathology. In: Javadpour N, ed. Principles and management of adrenal cancer. Berlin: Springer-Verlag, 1987;19-55). Focal chronic adrenalitis is common and has been found in up to 48% of autopsies, appearing as small aggregates of lymphocytes admixed with plasma cells adjacent to veins or venules in the corticomedullary junction.99 Focal chronic adrenalitis of this type is not considered to be a primary adrenal disorder but represents a nonspecific inflammatory reaction, possibly related to inflammation in neighboring organs, such as chronic pyelonephritis. Autoantibodies may be directed against adrenal medullary or chromaffin cells in type 1 (insulin-dependent) diabetes mellitus, and this organ-specific autoimmunity might be related to diabetic autonomic neuropathy.4 Members of the herpes virus group may infect the adrenal gland, including herpes simplex, cytomegalovirus, and varicella-zoster virus. A characteristic pattern of herpetic adrenalitis occurs with disseminated herpes simplex infection in newborns, referred to as neonatal hepatoadrenal necrosis and first described by Haas in 1935.100 The foci of herpes simplex or varicella-zoster infections tend to be small, circumscribed, punched-out areas of coagulative necrosis, with scant inflammation. The necrosis within the cortex may become widespread and confluent.3 Eosinophilic Cowdry type A intranuclear inclusions are the diagnostic hallmark, usually occurring in cells bordering the zones of necrosis (Fig. 16-17, A). It may not be possible to distinguish varicella-zoster infection from herpes simplex by routine light microscopy. Fig. 16-17 Herpetic adrenalitis. A, Herpes simplex virus infection. Note multiple intranuclear eosinophilic Cowdry type A inclusions (arrows) with peripheral displacement of nuclear chromatin (inset). B, Cytomegalovirus infection. The adrenal cortical cells infected by the virus show the characteristic cytomegalic changes. Insets highlight the large, amphophilic intranuclear inclusions with peripheral halo. Disseminated cytomegalovirus (CMV) infection in the newborn involves a wide variety of organs and tissues, including the adrenal gland. Some infants may have multiple mobile blue-gray subcutaneous nodules caused by dermal erythropoiesis. The appearance of the pigmented nodules may be striking, giving the infant an appearance described as a “blueberry muffin.”3 The viral cytopathic effect in CMV adrenalitis is virtually pathognomonic, with sharply defined large amphophilic intranuclear inclusions having characteristic halos and small, basophilic granules within the cytoplasm (see Fig. 16-17, B). CMV adrenalitis is relatively common in patients with AIDS; death is attributed in part in some cases to adrenal cortical insufficiency. In one study, approximately 50% of patients with AIDS had evidence of CMV infection at autopsy, and the adrenal glands were most commonly involved (75%) with cortical or medullary necrosis.101 Necrosis of the medulla caused by CMV infection may be greater than that of the cortex, and it is useful to note that there is no deficiency syndrome caused by destruction of the adrenal medulla. CMV infection of the adrenal in AIDS may result in latent or overt adrenal cortical insufficiency, requiring prophylactic treatment with corticosteroids.102 Rarely the adrenal is involved by other infectious agents, such as Pneumocystis jiroveci (Fig. 16-18), the most common opportunistic infection in patients with AIDS that rarely also occurs in immunocompetent individuals.103,104 Malakoplakia of the adrenal gland has been reported.105,106 Echinococcal cyst of the adrenal gland is usually an incidental autopsy finding.107 Fig. 16-18 A, Adrenal gland extensively involved by Pneumocystis jiroveci infection in a patient with acquired immunodeficiency syndrome. Note the characteristic foamy exudate in the cortex. B, Gomori methenamine silver stain shows several P. jiroveci organisms (arrow), some organisms have cup-shaped indentations when viewed in profile. Cortical nodularity in the adrenal can pose significant diagnostic problems for the pathologist at the autopsy table and in surgical material. Nodularity usually occurs in individuals without biochemical or clinical signs or symptoms of adrenal cortical hyperfunction. By definition, hyperplasia refers to an increase in the number of cells in tissue or in an organ, which in the adrenal cortex results in diffuse, symmetric thickening with or without cortical nodules (Fig. 16-19). The spectrum of diffuse and nodular adrenal cortical hyperplasia is broad. The incidence of cortical nodularity with eucorticalism can be analyzed from material obtained at autopsy or in patients who are discovered to have cortical nodules in vivo. Fig. 16-19 Schematic view of nodular adrenal gland in transverse section. Accessory nodules of cortical cells may be seen lying free within periadrenal fat, on the capsular surface, or attached to underlying cortex (capsular extrusion). A dominant macronodule within the cortex can simulate a neoplasm. Multiple small capsular arterioles are present on the surface of the gland. The central adrenal vein in the medullary compartment has discontinuous bundles of smooth muscle, allowing close contact of chromaffin cells or cortical cells with vascular lumina. Occasionally a mushroom-like intravascular protrusion by cortical or medullary cells can be seen.
Adrenal glands
Embryology and normal gross anatomy
Adrenal cortex

Adrenal medulla
Microscopic anatomy
Examination of the adrenal glands
Congenital and other abnormalities
Adrenal aplasia and hyperplasia
Adrenal heterotopia
Union and adhesion
Adrenal cytomegaly
Adrenoleukodystrophy
Congenital adrenal hyperplasia
Pathology of adrenal glands in congenital adrenal hyperplasia
Testicular tumors in congenital adrenal hyperplasia (testicular adrenogenital tumors)
Other tumors associated with congenital adrenal hyperplasia
Stress-related changes of the adrenal gland
Other abnormalities
Nonneoplastic diseases
Chronic adrenal cortical insufficiency (Addison disease)
Idiopathic or autoimmune Addison disease
Adrenal tuberculosis
Histoplasmosis and other fungal infections
Amyloidosis
Acute adrenal cortical insufficiency
Inflammation and other infections
Nonspecific adrenalitis
Herpetic adrenalitis
Rare infections
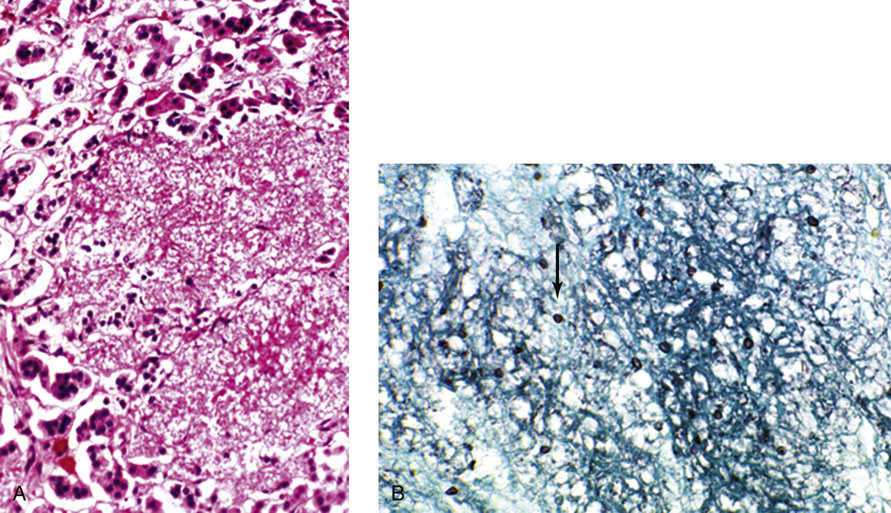
Adrenal cortical hyperplasia
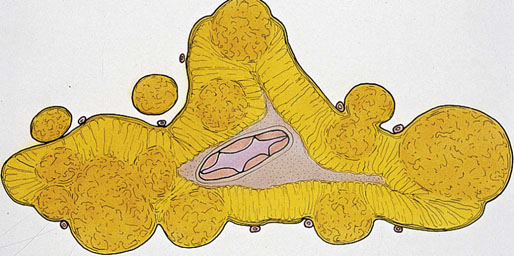
Nodular adrenal gland
Adrenal glands