CHAPTER 54 Adenocarcinoma and Other Tumors of the Stomach
EPIDEMIOLOGY
Gastric cancer remains the second leading cause of cancer mortality in the world,1 although the overall incidence is declining.2 The incidence of gastric cancer in Western countries has decreased dramatically over the past century.3 For example, gastric cancer mortality has decreased 86% since 1950 in the United States, and the incidence of gastric cancer has diminished four-fold since 1930 to approximately 8 cases per 100,000 people.4,5 As recently as 1930, gastric cancer was the leading cause of cancer mortality in the United States for men and the third leading cause for women.6 Gastric cancer is now the seventh leading cause of cancer mortality in the United States.5 It was estimated that in 2008, approximately 21,500 Americans would be diagnosed with gastric cancer and 10,880 would die of it.7
There is great geographic variation in gastric cancer incidence, with the highest incidence rates in the Far East (Fig. 54-1). Eastern Europe and Central and South America also have high incidence rates, and the lowest incidence rates are observed in North America, North Africa, South Asia, and Australia.6 Although gastric cancer was common in industrialized countries in the past, the latest epidemiologic data indicate that more than 60% of new cases of gastric cancer are in developing countries, reflecting a more rapid decline in developed countries.
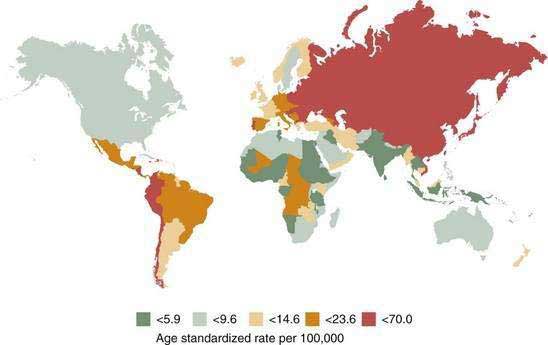
Figure 54-1. Worldwide incidence of gastric cancer in men.
(From Parkin DM. International variation. Oncogene 2004; 23:6239-40.)
In the United States, the median age of diagnosis is 71 years, with the highest proportion (28%) diagnosed between the ages of 75 and 84.8 In Japan, a country with a high incidence of gastric cancer, the mean age of diagnosis is roughly a decade earlier, perhaps reflecting lead-time bias due to widespread screening. The incidence of gastric cancer in men is approximately twice that in women (Table 54-1).3 The incidence of gastric cancer in blacks in the United States is nearly double that in whites.5 Native Americans and Hispanics also have a higher risk of development of gastric cancer than whites.9 Lower socioeconomic status is associated with a much higher incidence of gastric cancer.3 In the United States, the distribution of gastric cancer within the stomach is 39% in the proximal third, 17% in the middle third, 32% in the distal third, and 12% involving the entire stomach.10 In contrast to the pattern seen with noncardia tumors, the incidence rates of gastric cardia cancer are rising.2,11
Table 54-1 Gastric Cancer Incidence and Mortality Rates per 100,000 US Population by Race from 2001 to 2005
Dietary, environmental, and genetic risk factors for gastric adenocarcinoma are listed in Table 54-2, some of which are or may be protective.
Table 54-2 Risk Factors Including Protective Factors for Gastric Adenocarcinoma
| Definite | Helicobacter pylori infection |
| Chronic atrophic gastritis | |
| Intestinal metaplasia | |
| Dysplasia* | |
| Adenomatous gastric polyps* | |
| Cigarette smoking | |
| History of gastric surgery (esp. Billroth II)* | |
| Genetic factors | |
| Family history of gastric cancer (first-degree relative)* | |
| Familial adenomatous polyposis (fundic gland polyps)* | |
| Hereditary nonpolyposis colorectal cancer* | |
| Peutz-Jeghers syndrome* | |
| Juvenile polyposis* | |
| Probable | High intake of salt |
| Obesity (adenocarcinoma of cardia only) | |
| Snuff tobacco use | |
| History of gastric ulcer | |
| Pernicious anemia* | |
| Regular aspirin or NSAID use (protective) | |
| Possible | Low socioeconomic status |
| Ménétrier’s disease | |
| High intake of fresh fruits and vegetables (protective) | |
| High ascorbate intake (protective) | |
| Questionable | Hyperplastic and fundic gland polyps |
| High intake of nitrates | |
| High intake of green tea (protective) |
NSAID, nonsteroidal anti-inflammatory drug.
* Surveillance for cancer is suggested in patients with this risk factor.
ETIOLOGY AND PATHOGENESIS
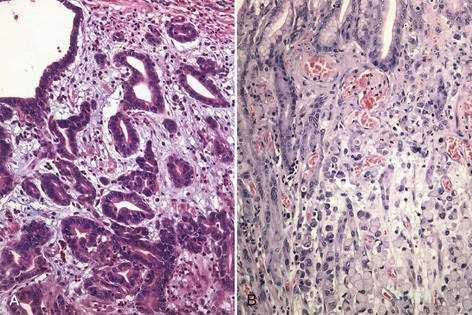
Gastric cancer can be subdivided using the Lauren classification into two distinct histologic subtypes with different epidemiologic and prognostic features (Fig. 54-2).12 The intestinal type of cancer is characterized by the formation of gland-like tubular structures with features reminiscent of intestinal glands. This type of gastric cancer is more closely linked to environmental and dietary risk factors, tends to be the predominant form in regions with a high incidence of gastric cancer, and is the form of cancer that is now declining worldwide. The diffuse type of cancer lacks glandular structure and consists of poorly cohesive cells that infiltrate the wall of the stomach. It is found at the same frequency throughout the world, occurs at a younger age, and is associated with a worse prognosis than the intestinal form. Extensive involvement of the stomach by the diffuse type can result in a rigid and thickened stomach, a condition referred to as linitis plastica. Adenocarcinoma of the stomach can also be classified into proximal tumors (gastric cardia and gastroesophageal junction) and distal tumors (fundus, body and antrum of the stomach). Distal tumors have been declining, whereas proximal tumors have been increasing (see Chapter 46).
It is now believed that the development of intestinal-type cancers occurs through a multistep process in which the normal mucosa is sequentially transformed into a hyperproliferative epithelium, followed by an early adenoma, late adenoma, and then carcinoma. In colon cancer, the evidence is strong that each step in the transition is associated with a specific gene mutation,13 but evidence that gastric cancer follows a comparable sequence of genetic events has been lacking. However, in gastric and colon cancer, it does appear that deoxyribonucleic acid (DNA) mutations are established over time in stem cells; in intestinal metaplasia these mutations spread through the stomach through a process involving crypt fission and monoclonal conversion of glands.14 The contention that the pathogenesis of intestinal-type gastric cancer is a multistep process is supported mainly by the observation that both atrophic gastritis and intestinal metaplasia are found in higher incidences in patients with intestinal-type cancer and in countries with a high incidence of gastric cancer (see Chapter 51).15
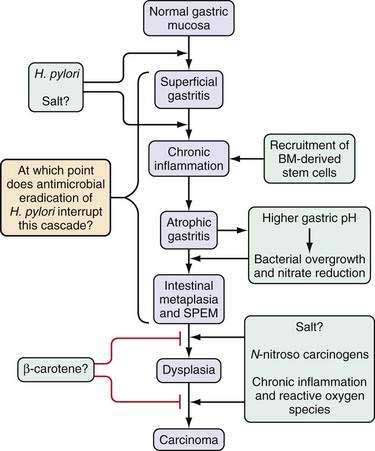
This multistep model of intestinal-type gastric cancer, developed in large part by Correa and colleagues,16,17 postulates that there is a temporal sequence of preneoplastic changes that eventually lead to the development of gastric cancer. A common feature of the initiation and progression to intestinal-type gastric cancer is chronic inflammation. Helicobacter pylori infection is the primary cause of gastric inflammation and the leading etiologic agent for gastric cancer (see Chapter 50). In a subset of patients infected with H. pylori, the inflammatory process leads to the development of atrophic gastritis (with loss of glandular tissue) followed by progression to intestinal metaplasia, dysplasia, early gastric cancer, and, eventually, advanced gastric cancer (Fig. 54-3). The current view is that all stages prior to the development of high-grade dysplasia are potentially reversible, although this concept is still somewhat controversial, it has been supported by a number of studies in animal models.18,19 Unlike the situation observed with colon cancer, the precise genes involved in each step of this progression are still not defined. Furthermore, during endoscopy the premalignant stages of gastric cancer are not as readily identifiable as those of colon cancer, and many gastric carcinomas are very heterogeneous, containing a large percentage of stromal cells. These stromal cells, which include cancer-associated fibroblasts known to promote tumor growth, have been reported to show distinct genetic and epigenetic changes that may confound tumor analysis.20 This feature makes characterization of the timing of specific gene mutations in gastric cancer difficult at best. Currently the role of chronic inflammation in the diffuse type of gastric cancer remains to be clarified, and the similarities if any to the proposed pathway in Figure 54-3 for the intestinal type of cancer.
HELICOBACTER PYLORI INFECTION
H. pylori is a gram-negative microaerophilic bacterium that infects nearly half of the world’s population and is recognized as the primary etiologic agent for gastric cancer (see Chapter 50). Indeed, H. pylori has been classified as a class I (or definite) carcinogen by the International Agency for Research on Cancer (IARC), a branch of the World Health Organization (WHO). Infection with H. pylori has been found in every population studied, although the prevalence is higher in developing countries.21,22
The natural history of chronic H. pylori infection includes three possible outcomes23: (1) superficial gastritis, in which most patients remaining asymptomatic; (2) duodenal ulcer phenotype, which occurs in 10% to 15% of infected subjects; and (3) gastric ulcer/gastric cancer phenotype, which is the least common in the United States. In general, the risk for gastric cancer is dependent on the types of gastritis, and an increased risk is associated with a low acid state. H. pylori–induced duodenal ulcer disease is associated with a high gastric acid output as well as a reduced risk for developing gastric cancer.24 Patients with H. pylori–associated gastric ulcer disease exhibit low gastric acid output, and their ulcers are typically associated with preneoplastic changes of atrophic gastritis and metaplasia. Overall, studies suggest that H. pylori–infected patients are at risk for development of chronic atrophic gastritis at a rate of 1% to 3% per year of infection.17,25,26 Thus, those patients who are genetically predisposed to forming atrophic gastritis in response to H. pylori infection are predisposed to gastric cancer. Although Helicobacter infection is associated with both intestinal-type and diffuse-type adenocarcinomas, the mechanisms responsible for the formation of intestinal-type adenocarcinoma have been better studied and are focused on here. The association of H. pylori with mucosa-associated lymphoid tissue (MALT) lymphoma is discussed briefly at the end of this chapter and in more detail in Chapter 29.
The increased risk of development of gastric adenocarcinoma due to H. pylori infection depends on multiple factors including the strain of bacteria, host genetic factors, the duration of infection, and the presence or absence of other environmental risk factors (e.g., poor diet, smoking, etc.). In a Japanese cohort of 1526 patients with peptic ulcer disease, nonulcer dyspepsia, and gastric hyperplasia, only those infected with H. pylori developed gastric adenocarcinoma during follow-up (2.9% vs. 0%, P < 0.001).27 Additional cohort studies from China and Taiwan have reported similar findings.28,29 At least in Western countries, the association between H. pylori and gastric cancer appears to be confined to noncardia gastric tumors.30
Potential mechanisms for H. pylori–induced gastric carcinogenesis include host factors, bacterial factors, environmental factors, and interactions among all three factors. Our latest understanding suggests that a combination of a virulent bacterial strain, a genetically permissive host, and a favorable gastric environment are necessary for disease to occur. The most important factor appears to be the induction of chronic inflammation by H. pylori infection. Several aspects of the inflammatory milieu have been implicated as carcinogens; they include increased oxidative stress and the formation of oxygen free radicals leading to DNA damage, increased CD4+ T cells and myeloid cells, and elevated proinflammatory cytokine production, all leading to accelerated cell turnover, reduced apoptosis, and the potential for faulty or incomplete DNA repair.31 Indeed, recent studies suggest that animals with deficient DNA repair mechanisms display more severe gastric dysplasia after chronic infection with H. pylori.32 Thus, evidence to date clearly indicates that the most important cofactor in the induction of Helicobacter-related disease is the host immune response. Indeed, chronic inflammation has been linked to a large number of cancers.
H. pylori infection leads to innate and adaptive immune responses (see Chapter 2). Initiation of the innate immune response to H. pylori is just beginning to be unraveled. Classically, the innate immune system consists of professional antigen presenting cells (APCs) such as macrophages, dendritic cells, and in some cases epithelial cells. Recent work supports a role for pattern recognition receptors (Toll-like receptors [TLRs]) in the initial response to Helicobacter colonization and the subsequent induction of the adaptive response. The most convincing evidence to date implicates TLR2 as the major TLR in Helicobacter species recognition.33 A role for TLRs 4, 5, and 9 remains more controversial.34–37 TLR4, along with CD14 and MD-2, serves as the receptor for Escherichia coli lipopolysaccharide (LPS) and probably H. pylori LPS and thus may be involved as well.
The C57BL/6 mouse is a susceptible inbred strain, in which initial colonization of the antrum by bacteria later spreads to the body or corpus, leading to severe chronic inflammation and increases in apoptosis (programmed cell death) and proliferation. The alterations in cellular turnover are associated with a loss of parietal and chief cells (atrophy), intestinal metaplasia, and dysplasia, followed by invasive gastric adenocarcinoma in mice 14 to 22 months after infection.38,39 Genetic manipulation of the C57BL/6-susceptible murine strain has facilitated detailed study and has thus led to a deeper understanding of genetic factors that promote murine gastric cancer, and in particular, the role of the adaptive immune response. For example, gastric Helicobacter infection in mice deficient in lymphocytes, including mice with recombinase-activating gene (RAG) deficiency, severe combined immunodeficiency, or T cell deficiency, does not result in tissue damage, cell lineage alterations, or the metaplasia-dysplasia-carcinoma sequence.40,41 Infection in B cell–deficient mice (which retain a normal T cell response) results in severe atrophy and metaplasia identical to that seen in infected wild-type mice.41 Taken together these studies underscore the crucial role of CD4+ T lymphocytes in orchestrating gastric neoplasia.
How do CD4+ T lymphocytes contribute to gastric cancer progression? Susceptible mouse strains, such as C57BL/6, mount a strong helper T cell type 1 (Th1) interferon-γ (IFN-γ), interleukin-12 (IL-12) type of immune response, whereas resistant strains, such as the BALB/c, have an opposite Th2 response (IL-4, IL-5).39,42,43 A Th2 response is associated with protection from mucosal damage despite the inability to eliminate bacterial colonization and in fact is often associated with higher bacterial colonization rates. Mouse strains such as the C3H, which has a mixed Th1/Th2 cytokine profile, show intermediate disease, suggesting that cytokines within an immune response interact to form a continuum of disease rather than discrete disease states. More recently, Th17 cells, which express IL-17, have been shown to be an important component of H. pylori–induced gastritis.
Although the composite immune milieu most likely dictates disease manifestations, studies are beginning to define the role of individual cytokines in the predisposition to disease. This is best illustrated in the IFN-γ knockout mice, in which a lack of IFN-γ protects infected mice from atrophy.39,43 On the other hand, mice lacking IL-10, a cytokine that acts to dampen an immune response, develop severe atrophic gastritis in response to infection.39–43 More recently, genetic murine models have illustrated the importance of the IL-6–IL-11 family of cytokines in the development of gastric cancer.44
Manipulation of the immune response within wild-type strains confirms the central role of the Th1/Th2 response in producing disease. For example, infection with the intestinal helminth Heligmosomoides polygyrus skews the immune response toward Th2 polarization and protects the C57BL/6 host from Helicobacter-induced atrophy and metaplasia.45 This mouse model mimics both the parasitic infection status and the paradoxical low gastric cancer–high H. pylori infection rates seen in areas of Africa, potentially explaining this apparent inconsistency. These observations in mice led to human studies in Africa and Latin America that confirmed that geographic regions with low gastric cancer rates had much higher Th2 relative to Th1 immune responses to H. pylori.46,47 In general, the increased Th2-type responses were found in areas where serum immunoglobulin E (IgE) levels were high and the prevalence of intestinal parasitism by helminths is greater than 50%. These findings further stress the importance of the host response to infection and suggest the possibility that manipulation of the genetically predetermined host cytokine profile in response to environmental challenges may lessen or exacerbate the disease process.
There is a great deal of genetic diversity between strains of H. pylori owing to point mutations, insertions, deletions, and base-pair substitutions within its genome. Several strains may infect a single individual, and existing strains can undergo mutations and change over time.48,49 Despite this genetic diversity, several genes are recognized as risk factors for gastric carcinoma, including the cag pathogenicity island, the vacA gene, and the babA2 gene.
The H. pylori genome is 1.65 million base pairs and codes for approximately 1500 genes, two thirds of which have been assigned biological roles.50 The function of the remaining one third of the genome remains obscure. Factors that contribute to carcinogenesis include those that enable the bacteria to effectively colonize the gastric mucosa, those that incite a more aggressive host immune response, and those that affect host cell-growth signaling pathways.
Motility toward epithelial cells of the stomach is a vital feature of H. pylori survival tactics. This function is ensured by several factors, including spiraling movement (FlaA and FlaB proteins), which are designed to navigate the thick gastric mucus and through efficient modifications of the extracellular matrix and mucus layer, thus decreasing viscosity and allowing bacterial penetration.51,52 In addition, H. pylori expresses a variety of genes that contribute to buffering of stomach acid in order to maintain a relatively neutral pH. This includes a urease gene cluster, consisting of seven genes, of which UreA/UreB complex (comprising the urease enzyme) codes for 10% of the protein of H. pylori and is vital for its survival.
A significant proportion (e.g., ≈20%) of H. pylori organisms can be found adherent to the surface of gastric mucous cells. Occasionally H. pylori can also be found intracellularly, particularly in preneoplastic and neoplastic lesions.53 Adhesion of the bacteria to the epithelial layer is facilitated by a large family of 32 related outer-membrane proteins (Hop proteins) that include the adhesins. One of the best-characterized adhesins is BabA, which is encoded by the strain-specific gene babA2, a member of a highly conserved family of outer membrane proteins. BabA binds to the fucosylated Lewis B blood group antigen on gastric epithelial cells and forms a scaffold apparatus that allows bacterial proteins to enter host epithelial cells. Bacterial strains that possess the babA2 gene adhere more tightly to epithelial cells, promote a more aggressive phenotype, and are associated with a higher incidence of gastric adenocarcinoma.54
The cag pathogenicity island is approximately 40 kb and contains 31 genes. The terminal gene of this island, cagA, is often used as a marker for the entire cag locus. Compared with cagA-negative (cag−) strains, cag-positive (cagA+) strains are associated with more severe inflammation, higher degrees of atrophic changes, and a greater chance of progressing to gastric adenocarcinoma.55–58 The estimated risk has ranged from an odds ratio of 2 to as high as 28.4.23 However, many of the genes adjacent to cagA code for a type 4 secretion system (TFSS), often viewed as a molecule needle that injects bacterial proteins (such as CagA) into host cells. The remarkable finding that CagA is injected into host cells, where it is phosphorylated by Src and c-Abl kinases, has raised the possibility that CagA could directly promote growth, migration, and transformation. Indeed, transgenic expression of H. pylori CagA induces gastrointestinal (GI) and hematopoietic neoplasms in mice.59 Other genes within the pathogenicity island are also believed to be important for disease (cagE or picB, cagG, cagH, cagI, cagL, cagM) because they appear to be required for in vitro epithelial cell cytokine release, although they do not seem to have as great an effect on immune cell cytokine activation as cagA.60–62 These findings may explain the attenuated inflammatory response and lower cancer risk with cagA− strains in vivo.63–66
All strains of H. pylori carry the vacA gene, which codes for a pore-forming vacuolating toxin, but expression of vacA differs according to allelic variation. Approximately 50% of H. pylori strains express the vacA protein, which has been shown to be a very powerful inhibitor of T cell activation in vitro.67 Although vacA and cagA map to different loci within the H. pylori genome, the vacA protein is commonly expressed in cagA+ strains. There are various forms of vacA, and the s1m1 strains are highly toxigenic. Other bacterial virulence factors, such as cagE, may play a role in the modulation of apoptosis and the host inflammatory response, thereby contributing to disease manifestations. Indeed, “virulent strains” (cagA+, cagE+, and VacA+ s1m1) appear to be more potent inducers of proinflammatory mediators than “nonvirulent strains” (cagA−, cagE−, and VacA−), possibly explaining the higher association of cagA+ strains with gastric cancer.68
DIETARY FACTORS
Numerous dietary factors have been implicated as risk factors for gastric cancer. The decline in gastric cancer rates has coincided with the widespread use of refrigeration and the concomitant higher intake of fresh fruits and vegetables and lower intake of pickled and salted foods. Use of refrigeration for more than 10 to 20 years has been associated with a decreased risk of gastric cancer.17,69 Lower temperatures reduce the rate of bacterial, fungal, and other contaminants of fresh food, as well as the bacterial formation of nitrites. Additionally, high intake of highly preserved foods may be associated with increased gastric cancer risk,70 likely because of higher contents of salt, nitrates, and polycyclic aromatic amines.71
Much attention has been given to the effects of high nitrate intake. When nitrates are reduced to nitrite by bacteria or macrophages, they can react with other nitrogenated substances to form N-nitroso compounds that are known mitogens and carcinogens.72,73 In rats, N-nitroso compounds have been shown to cause gastric cancer.74 However studies trying to link N-nitroso exposure to gastric cancer risk have been inconclusive, perhaps reflecting the fact that nitrate intake does not necessarily correlate with nitrosation levels.75,76 A Swedish cohort study found a nearly two-fold increased risk of gastric cancer associated with high dietary nitrate intake.70 However, separate large cohort studies from Europe did not demonstrate an association between nitrate intake and risk of gastric cancer.77,78
Another factor implicated in the development of gastric cancer is a diet high in salt (pickled foods, soy sauce, dried and salted fish and meat). High salt intake has been associated with higher rates of atrophic gastritis in humans and animals in the setting of Helicobacter infection79 and increases the mutagenicity of nitrosated food in animal models.17 High salt diets are associated with a roughly two-fold increased risk of gastric cancer.80,81 Cohort and case-control studies have also found an increased risk of gastric cancer associated with processed meat intake.70,82 Possible mechanisms include higher bacterial loads, up-regulation of H. pylori cagA expression, and increased cell proliferation and p21 expression.79,83,84
Epidemiologic studies have had inconsistent findings with regard to fruit and vegetable consumption and risk of gastric cancer. A cohort study from Japan found significantly decreased risks of gastric cancer associated with increased vegetable and fruit intake.85 A Swedish cohort study demonstrated a reduced risk of gastric cancer associated with high vegetable intake, but no association was seen with amount of fruit consumption.86 A large cohort study of nearly 500,000 adults in the United States and a separate nested case-control study from Europe failed to find an association between fruit and vegetable intake and gastric cancer risk.87,88
Other foods or dietary factors that have been implicated as potential risk factors for gastric cancer are high intake of fried food, foods high in fat, high intake of red meat, and aflatoxins.82,89–91 Diets with a high intake of fresh fish and antioxidants may be protective.90,92–94 However, there are insufficient data to make definitive conclusions regarding these factors.
CIGARETTE SMOKING
Tobacco has long been established as a carcinogen, and numerous epidemiologic studies have demonstrated an association between cigarette smoking and risk of gastric cancer.95 Several large cohort studies from Europe and Asia have reported a significantly increased risk of gastric cancer among smokers.96–98 A meta-analysis found that compared with never smokers, current smokers had a 1.5-fold increased risk of gastric cancer for the cardia as well as noncardia region.99 The authors also reported an increased association with greater amounts of smoking.
Moist snuff is a smokeless tobacco product promoted as an alternative to cigarettes and has reportedly reduced levels of carcinogenic nitrosamines. However, results of a Swedish cohort study demonstrated a 1.4-fold increased risk of noncardia gastric cancer among regular snuff users.100 Snuff exposure also increases the rate of gastric carcinogenesis in H. pylori–infected mice.101
ALCOHOL
Multiple cohort and case-control studies from the United States and Europe have found no significant association between alcohol consumption and cardia or noncardia gastric cancer.98,102,103 A separate population-based case-control study in the United States also found no association between any alcohol use and risk of either cardia or noncardia gastric cancer, although a reduced risk was reported with wine consumption (cardia, odds ratio [OR] 0.6; 95% confidence interval [CI]: 0.5 to 0.9; noncardia, OR 0.7; 95% CI: 0.5 to 0.9).104
OBESITY
Obesity is a recognized risk factor for numerous gastrointestinal malignancies.105 Increased body mass index (BMI) appears to be associated with a mild to moderate increased risk of gastric cardia cancer but not for noncardia gastric cancer.106–110 Results of the National Institutes of Health–American Association of Retired Persons (NIH-AARP) Diet and Health Cohort Study demonstrated that marked obesity (BMI = 35 kg/m2) was associated with a significantly increased risk of gastric cardia cancer (hazard ratio, 2.46) but not with noncardia gastric cancer.106 A separate cohort study from the Netherlands also found an increased risk of cardia cancer with increasing BMI.107
INHERITED PREDISPOSITION
Overall, 10% of cases of gastric cancer appear to exhibit familial clustering,111 and family history is likely an independent risk factor for the disease even after controlling for H. pylori status.112,113 In a cohort study of relatives of patients with gastric cancer, siblings had a two-fold increased risk of gastric cancer, adjusted for H. pylori infection.114 In a case-control study from Japan, a positive family history was associated with a significantly increased odds of gastric cancer in women (OR, 5.1), but not in men.115 A study from Scandinavia showed that having a twin with gastric cancer conferred a markedly higher risk for the disease (hazard ratios, 9.9 for monozygotic twins and 6.6 for dizygotic twins), leading the researchers to calculate that heritable factors accounted for 28% of gastric cancers, compared with 10% for shared environmental factors and 62% for nonshared environmental factors.116
Some of the familial clustering seen with intestinal-type gastric cancer may be related to genetic factors that play a role in the host immune response to H. pylori infection. Data from South Korea indicate that individuals with a family history of gastric cancer more frequently have H. pylori infection as well as associated atrophic gastritis or intestinal metaplasia.117 In a case-control study from Scotland, relatives of patients with gastric cancer had a higher prevalence of atrophy and hypochlorhydria, but a similar prevalence of H. pylori infection, compared with controls.118 The greater prevalence of atrophy was confined to those patients with H. pylori infection, suggesting the possibility these individuals were perhaps exhibiting a more vigorous immune response to H. pylori. In a number of model systems, the development of gastric atrophy has been linked to a strong Th1 immune response.43,45,119 Thus, it was postulated that candidate disease-susceptibility genes for gastric atrophy and cancer might be genes that participate in the innate and adaptive immune responses to H. pylori infection. Inflammation is modulated by an array of pro- and anti-inflammatory cytokines, and several genetic polymorphisms have been described that influence cytokine response.
IL-1β is an important proinflammatory cytokine and a powerful inhibitor of acid secretion. Thus, the initial report in this area described in the setting of H. pylori infection an association between proinflammatory IL-1 gene cluster polymorphisms (IL-1B encoding IL-1β, and IL-1RN encoding its naturally occurring receptor antagonist, IL-1RA) and neoplastic progression. Individuals with the IL-1β-31*C or -511*T and IL-1RN*2/*2 genotypes were shown in the study to be at higher risk for development of H. pylori–dependent hypochlorhydria and gastric cancer.120 The increased risk of progression to cancer with these genotypes was in the two- to three-fold range compared with noninflammatory genotypes. The initial report was confirmed in other studies.121–125 Subsequently, Hwang and colleagues126 demonstrated that carriers of the IL-1β-511T/T genotype or the IL-1RN*2 allele had higher mucosal IL-1β levels than noncarriers, and they also confirmed the association between the -511T/T genotype and severe gastric inflammation and atrophy. The importance of IL-1β in carcinogenesis has now been demonstrated in a transgenic study, in which stomach-specific expression of human IL-1β in transgenic mice led to spontaneous gastric inflammation and cancer that correlated with early recruitment of myeloid-derived suppressor cells (MDSCs) to the stomach.127
Additional associations with gastric cancer risk have been reported for genetic polymorphisms in tumor necrosis factor-α (TNF-α) and IL-10. Proinflammatory genotypes of TNF-α and IL-10 each were associated with a two-fold higher risk of noncardia gastric cancer. When combined with proinflammatory genotypes of IL-1β and IL-1RN, patients with three or four high-risk genotypes showed a 27-fold greater risk of gastric cancer.128 Additional studies have shown that polymorphisms of the TLR4 gene also increase the risk of gastric cancer. Carriers of the TLR4+896G polymorphism had an 11-fold increased risk of hypochlorhydria, and significantly more severe gastric atrophy and inflammation.129 Accumulated evidence suggests that the genetic predisposition to gastric cancer may be largely determined by the TLR and cytokine responses to chronic Helicobacter infection.
The best described form of hereditary gastric cancer is the diffuse gastric cancer that is seen in the presence of a germline mutation in the gene CDH1, which encodes the cell adhesion molecule E-cadherin. A large New Zealand kindred was found to have a germline mutation in the E-cadherin gene, and similar mutations have been reported in several additional kindreds, all with diffuse-type gastric cancer.130–133 The age of onset of gastric cancer in individuals with CDH1 mutations is less than 40 years but can be highly variable, and the estimated lifetime risk of gastric cancer is close to 70%.134,135 Germline CDH1 mutations are also associated with familial lobular breast cancer.136,137
A small part of the familial clustering of gastric cancer can be attributed to other cancer syndromes (see Chapter 122). Patients with familial adenomatous polyposis (FAP) have a prevalence of gastric adenomas ranging from 35% to 100%, and their risk of gastric cancer is close to 10-fold higher than that of the general population.138 These cancers frequently arise from fundic gland polyps and develop at an early age.139,140 Patients with hereditary nonpolyposis colorectal cancer (HNPCC) syndrome have an approximately 11% risk of developing gastric cancer, predominantly of the intestinal type, with a mean age at diagnosis of 56 years.141 Patients with juvenile polyposis also have a 12% to 20% incidence of gastric cancer.142,143
GENETICS
Aneuploidy is common in gastric cancer (seen in 60% to 75% of cases), but cytogenetic studies have failed to identify any consistent chromosomal abnormality. Comparative genomic hybridization studies have shown that chromosome arms 4q, 5q, 9p, 17p, and 18q exhibit frequent decreases in DNA copy number, whereas chromosomes 8q, 17q and 20q often have increased DNA copy number.144
There is a consensus that TP53 is the most commonly mutated gene in gastric cancer (60% to 70% of gastric cancers) and that mutations in Ras, APC, and Myc are rare.145,146 Loss of heterozygosity (LOH) at the APC locus occurs more commonly. Another genetic abnormally found at high frequency includes deletion or suppression of the fragile histidine triad gene (FHIT) (60%), a tumor suppressor locus on chromosome 3p. Genes that normally inhibit entry into the cell cycle, such as p16 and p27, show diminished expression in nearly one half of gastric cancers.147–152 Absence of p27 expression is associated with a poorer prognosis.147,149 Absence of p16 expression is seen most commonly in poorly differentiated carcinomas but has no measurable impact on patient prognosis.153 Diminished expression of p16 and p27 occurs in the absence of detectable mutations and is believed to be secondary to hypermethylation of the respective genes.151 Many of these cancers show hypermethylation of a number of promoter regions, including the MLH1 promoter region, and show the high-level microsatellite instability (MSI) phenotype (see Chapter 3). Multiple tumor suppressor genes have been shown to be methylated in gastric cancers. Emerging evidence suggests that epigenetic changes, including global hypomethylation and promoter hypermethylation, occurs quite early in gastric carcinogenesis. In addition, it appears that DNA methylation changes also occur in the tumor-associated stromal fibroblasts, suggesting an important role for the tumor microenvironment.20
Overexpressions or amplifications of a number of growth factor pathways have been described, including cyclooxygenase-2 (COX-2) (70%), hepatocyte growth factor/scatter factor (HGF/SF) (60%), vascular endothelial growth factor (VEGF) (50%), c-met (50%), amplified in breast cancer-1 (AIB-1) (40%), and β-catenin (25%) (Table 54-3).154 Approximately 15% of gastric cancers have been reported to overexpress both epidermal growth factor (EGF) and EGF receptor (EGFR), consistent with an autocrine mechanism. Mutations in PIC3A, a gene that codes for a catalytic subunit of phosphotidylinositol 3-kinase (PI3K), has been found in up to 25% of gastric cancers analyzed.155 In addition, mutations in human protein tyrosine phosphatases (PTPs) were found by the same laboratory in 17% of gastric cancers, with the protein tyrosinase phosphatase–receptor type (PTPRT) the most frequently altered.156
Table 54-3 Genetic Abnormalities in Gastric Adenocarcinoma
| ABNORMALITIES | APPROXIMATE GENE FREQUENCY (%) |
|---|---|
| DNA aneuploidy | 60-75 |
| Microsatellite instability | 15-50 |
| Deletion/Suppression | |
| TP53 gene | 60-70 |
| Fragile histidine triad gene (FHIT) | 60 |
| Adenomatous polyposis coli (APC) gene LOH | 50 |
| Deleted in colorectal cancer (DCC) gene LOH | 50 |
| Decreased Expression Due to Hypermethylation | |
| p16 | ≈50 |
| TFF1 | ≈50 |
| p27 | <50 |
| MLH1 | 15-20 |
| E-cadherin | 50 |
| Amplification/Overexpression | |
| Cyclooxygenase-2 (COX-2) | 70 |
| Hepatocyte growth factor (HGF) | 60 |
| Vascular endothelial growth factor (VEGF) | 50 |
| c-Met | 50 |
| Amplified in breast cancer-1 (AIB-1) | 40 |
| Beta-catenin | 25 |
| EGF/EGFR | 15 |
| Mutations | |
| PI3K | 25 |
| PTPRT | 17 |
DNA, deoxyribonucleic acid; EGF, epidermal growth factor; EGFR, epidermal growth factor receptor; LOH, loss of heterozygosity; MLH1, human mutL homolog 1; PI3K, phosphatidylinositol 3-kinase; PTPRT, protein-tyrosine phosphatase receptor-type; TFF1, human trefoil factor 1.
Gastric-specific tumor suppressor genes TFF1 (Trefoil factor 1) and RUNX3 (Runt-related transcription factor 3), which have now been identified and may represent “gatekeepers” of the gastric cancer pathway, are logical targets for further study.157,158 Loss of TFF1 has been described in around 50% of gastric carcinomas, and TFF1 knockout mice develop spontaneous gastric antral tumors. Mutations of TFF1 also have been described, and these enhance gastric cancer cell invasion through signaling pathways that include PI3-K and phospholipase-C.159 TFF1 expression is repressed by STAT-3, and activation of STAT-3 is also emerging as a key pathway that leads to gastric cancer.44 RUNX3 most likely suppresses gastric epithelial growth by inducing p21 and Bim, attenuating Wnt signaling, and is altered in 82% of gastric cancers.160 Investigations into these genes and their contributions to the gastric cancer phenotype will prove valuable to our understanding of disease progression.
MSI in dinucleotide repeats secondary to defects in DNA mismatch repair genes, such as MLH1 and MLH2 (mutL homologs 1 and 2), have been implicated in the development of colorectal cancer, and in particular the HNPCC syndrome. Patients with HNPCC have an 11% incidence of gastric cancer, suggesting that MSI may also play a role in the development of gastric cancer.141 MSI is found in 15% to 50% of sporadic gastric cancers, with a higher prevalence in the intestinal type of cancer.161–166 Low-level microsatellite activity (e.g., MSI-low) can be found in 40% of areas of intestinal metaplasia in patients with gastric cancer166 and in 14% to 20% of adenomatous polyps.164,166,167 MSI-high (MSI-H) occurs in only 10% to 16% of gastric cancers. MSI is associated with the less frequent occurrence of TP53 mutations, well- to moderately well differentiated histology, and distal tumor location. Studies that have examined the effect of MSI on patient survival have shown inconsistent results.167,168 When the findings are taken together, it would appear that MSI does play a role in the pathogenesis of gastric cancer, likely before the development of intestinal metaplasia (see Fig. 54-3), and is most commonly due to methylation of the MLH1 promoter.
The data regarding the genetics of diffuse gastric cancer are less complete. Mutations in the E-cadherin (CDH1) gene have been linked to the development of the diffuse type of gastric cancer. Several kindreds, families with hereditary diffuse gastric cancer (HDGC) have been found to carry a germline mutation in the CDH1 gene, all with diffuse-type cancer.130–132,169,170 Further evidence supporting a role for E-cadherin in the pathogenesis of gastric cancer comes from studies showing that suppression of E-cadherin expression occurs in 51% of cancers, with a higher percentage found in diffuse-type cancers.171 Furthermore, E-cadherin underexpression is associated with higher rates of lymph node metastases and reduced survival.172,173 The overall rates of CDH1 mutations in gastric cancer are low, however, with the decreased expression of E-cadherin seen in gastric cancer likely secondary to hypermethylation of the CDH1 promoter, which occurs in 50% of gastric cancers and 83% of diffuse-type gastric cancers.174 E-cadherin is a transmembrane protein that connects to the actin cytoskeleton through α- and β-catenins to establish cell polarity and mediates homophilic cellular interactions.175,176 Decreased expression of E-cadherin is believed to promote dissociation of cancer cells from their cell matrix, enhancing the migration and invasion of gastric cancer cells. Expression of α-catenin is also decreased or absent in 68% of gastric cancers.177 Therefore, E-cadherin appears to act as a tumor suppressor gene that may be important in the pathogenesis of diffuse gastric cancer.
Perhaps as important as the genetic alterations acquired during the progression to gastric adenocarcinoma, is in what target cells do these changes occur? In order for a cell to accumulate the quantity of genetic changes necessary for autonomous growth, it must be long lived. For these reasons, the current thinking is that a resident tissue stem cell is the target of genetic mutations and becomes the “cancer stem cell” capable of autonomous growth and with metastatic potential. Work from our laboratory offers a new model for the cancer stem cell. Bone marrow–derived stem cells are capable of homing to injured and inflamed peripheral organs and differentiating into organ-appropriate cell lineages.178–181 In an environment of inflammation and altered growth signaling, these cells can differentiate aberrantly and become dysplastic and neoplastic, and we have shown they constitute the majority of cells within in situ as well as invasive gastric adenocarcinoma lesions.182 Although much work needs to be done to understand these findings completely, they offer an exciting possibility for new approaches to understanding and treating gastric and other inflammatory mediated cancers.
PREMALIGNANT CONDITIONS
CHRONIC ATROPHIC GASTRITIS
Chronic atrophic gastritis, which is defined as the loss of specialized glandular tissue in its appropriate region of the stomach, is an established early morphologic change that occurs along the sequence toward the development of gastric cancer.16,183 The presence of atrophic gastritis has an annual incidence of progression to gastric cancer of approximately 0.5% to 1%.184–187 The extent of atrophic gastritis within the stomach correlates with risk of progression to cancer.188–190
There are two forms of atrophic gastritis. The more common is multifocal atrophic gastritis (MAG), which is associated with H. pylori infection and more likely to be associated with metaplasia. The presence of H. pylori infection is associated with an approximately 10-fold increased risk of atrophic gastritis.191 There is considerable regional variation in the prevalence of atrophic gastritis in H. pylori–infected individuals, with a roughly 3-fold increase in Asian compared with Western countries.191,192 The second form of atrophic gastritis, corporal atrophic gastritis, is associated with antiparietal cell and intrinsic factor antibodies. This form of atrophy is confined to the body and fundus. Corporal atrophic gastritis is associated with pernicious anemia and an increased gastric cancer risk, albeit not as high as that seen with H. pylori–induced multifocal atrophic gastritis, owing most likely to a lesser degree of inflammation.185,193
Mechanisms underlying the increased risk of gastric cancer in the setting of gastric atrophy may be related to low acid output (achlorhydria), which predisposes to gastric bacterial overgrowth (with non-Helicobacter organisms), greater formation of N-nitroso compounds, and diminished ascorbate secretion into the gastric lumen.194 Additionally, serum gastrin levels are increased in response to the reduced acid output. Gastrin is a known growth factor for gastric mucosal cells, and sustained elevations of serum gastrin may contribute to abnormal growth and increased risk of neoplastic progression.195,196
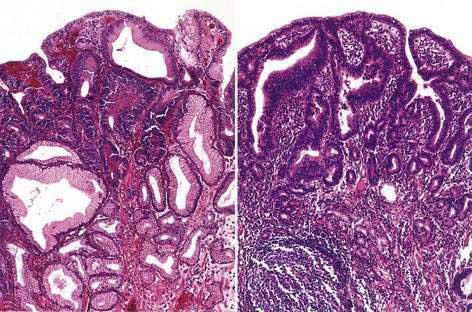
INTESTINAL METAPLASIA
Intestinal metaplasia (IM) can be subdivided into three categories, as classified by Jass and Filipe and as discussed in Chapter 51.197 Type I is the complete form of IM, containing Paneth cells, goblet cells that secrete sialomucins, and absorptive epithelium with well-defined brush borders. Type I metaplasia does not raise the risk of gastric cancer. Type II or incomplete metaplasia contains few absorptive cells, few columnar intermediate cells, and goblet cells that express sialomucins. Type III is intermediate between type I and type II and contains properties of both.198 It is estimated that the presence of type II or III IM is associated with a 20-fold increased risk of gastric cancer. Early gastric cancer develops in 42% of patients with type III IM within five years of follow-up, suggesting that IM represents a precursor lesion for the intestinal form of gastric cancer.199 However, whether cancer arises from areas of IM or whether IM simply represents a marker for higher gastric cancer risk remains unclear. As is the case with atrophic gastritis, the prevalence of IM in H. pylori–infected individuals is higher in Asia (≈40%) as compared with the West.191,192
DYSPLASIA
Histologic assessment of gastric dysplasia and adenocarcinoma is based on the 2000 Vienna classification (Table 54-4).200 The prevalence of gastric dysplasia ranges from as low as 0.5% in low-risk areas201 to 20% in high-risk areas.202 Prospective studies have shown that low-grade dysplasia may regress in up to 60% of cases, whereas it progresses to high-grade dysplasia in 10% to 20% of cases (Fig. 54-4).203–205 High-grade dysplasia rarely regresses, and is associated with an annual incidence of progression to gastric cancer of 2% to 6%.205–207 In a prospective cohort study from the Netherlands, the presence of high-grade dysplasia was associated with a markedly increased risk of progression to cancer (adjusted hazard ratio, 40.1).206 High-grade dysplasia is often associated with synchronous cancer and can be uni- or multifocal.208
Table 54-4 Padova International Classification System for Gastric Dysplasia
| CATEGORY | DEFINITION | HISTOLOGIC DESCRIPTION |
|---|---|---|
| I | 1.0 Normal | Normal gastric architecture with absent or minimal inflammatory infiltrates. |
| 1.1 Reactive foveolar hyperplasia | The general architecture is well preserved, with evidence of hyperproliferative epithelium, enlarged nuclei, and mitotic figures. | |
| 1.2 Intestinal metaplasia | Type I. Closely resembles the morphology of the small intestine, with absorptive enterocytes, well-defined brush borders, and well-formed goblet cells. | |
| Type II. Incomplete metaplasia with irregular mucous vacuoles, absence of brush borders, and difficult-to-identify absorptive enterocytes. Cells secrete mainly sialomucins. | ||
| Type III. Same as type II except cells secrete mainly sulfomucins. | ||
| II | Indefinite for dysplasia | Inability to discern whether cells are neoplastic or non-neoplastic. Usually found in setting of inadequate biopsy specimens and presence of architectural distortion and nuclear atypia. |
| III | Noninvasive neoplasia | Phenotypically neoplastic epithelium confined to glandular structures inside the basement membrane. Includes adenomas. |
| Should be divided into “low grade” and “high grade.” | ||
| IV | Suspicious for invasive cancer | Presence of neoplastic epithelium, but where invasion cannot be clearly identified. |
| V | Invasive cancer | Invasive carcinoma. |
Adapted from Rugge M, Correa P, Dixon M, et al. Gastric dysplasia: The Padova International Classification. Am J Surg Pathol 2000; 24:167.
GASTRIC POLYPS
The prevalence of gastric polyps in the general population is approximately 0.8% to 2.4%.209,210 Gastric polyps consist predominantly of fundic gland polyps (≈50%), hyperplastic polyps (≈40%), and adenomatous polyps (≈10%).210,211 The clinical course of fundic gland polyps is generally benign, and they are detected with increasing frequency in the era of proton pump inhibitor (PPI) use. In a series of 599 consecutive patients who underwent upper endoscopy, use of PPIs for 5 years or longer was associated with a significantly increased risk of fundic gland polyps (hazard ratio, 3.8).212 The rate of malignant transformation of fundic gland polyps is generally quite low (≈1%) and confined to polyps larger than 1 cm.213 One notable exception to the benign nature of fundic gland polyps is in patients with FAP. In this group the prevalence of fundic gland polyps ranges from 51% to 88%, with dysplasia present in more than 40% of cases.139,140
Hyperplastic polyps are almost always benign lesions. The rare hyperplastic polyps that undergo malignant transformation often contain areas of intestinal metaplasia or dysplasia and typically form a well-differentiated intestinal-type cancer.213
In contrast to fundic gland and hyperplastic polyps, gastric adenomas undergo malignant transformation at a high rate. Gastric adenomas that were followed by serial endoscopy with biopsy were documented to progress to dysplasia and then to carcinoma in situ, which developed within 4 years of follow-up in approximately 11% of cases.214
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


