Metabolic Alkalosis
Metabolic alkalosis is an acid–base disorder in which a primary disease process leads to the net accumulation of base within or the net loss of acid from the extracellular fluid (ECF). When it occurs as a simple acid–base disorder, it is recognized as an increase in plasma [HCO3−] and a compensatory increase in arterial blood pH.
Unopposed by other primary acid–base disorders, the increase in arterial blood pH promptly and predictably depresses ventilation resulting in increased Paco2 and buffering of the magnitude of the alkalemia; an increase in Paco2 of 0.6–0.7 mm Hg for every 1.0 mEq/L increase in plasma [HCO3−] is predicted from empirical studies. Although a Paco2 greater than 55 mm Hg is uncommon, compensatory increases to 60 mm Hg or higher have been documented in severe metabolic alkalosis. The magnitude of the compensatory increase in Paco2 is directly related to the extent of the alkalosis and the degree of elevation of the plasma [HCO3−], whether or not there is an intracellular acidosis. This compensatory respiratory response is independent of hypoxia, hypokalemia, renal failure, and cause and is usually not detectable clinically because it is more dependent on a change in depth rather than the rate of ventilation.
The major clinically and pathophysiologically relevant classification is based on whether the metabolic alkalosis is dependent on Cl− depletion. The Cl−-depletion forms, also termed the Cl−-responsive alkaloses, are more common. The other major grouping is the Cl−-resistant alkaloses, most of which are due to K+ depletion with mineralocorticoid excess. Mixed K+ and Cl−-depletion metabolic alkaloses also occur. Several other relatively uncommon causes constitute the balance of etiologies of metabolic alkalosis (Table 5–1).
Cl− depletion |
Gastrointestinal losses: Vomiting or nasogastric aspiration (even with achlorhydria), congenital chloridorrhea, gastrocystoplasty, villous adenoma |
Renal losses: Chloruretic diuretics, posthypercapnia, severe K+ depletion |
Skin losses: Cystic fibrosis |
K+ depletion |
Gastrointestinal losses: Laxative abuse |
Renal losses: Hyperaldosteronism, primary and secondary, other hypokalemic hypertensive syndromes, Bartter’s and Gitelman’s syndromes |
Miscellaneous |
Low glomerular filtration rate with base loading |
Milk-alkali syndrome |
Hypercalcemia |
Nonreabsorbable antacids with cation exchange resin |
Hypoalbuminemia |
Recovery from starvation |
Transient |
Multiple blood transfusions with citrate |
Infant formulas with low Cl− content |
Metabolic alkalosis should be anticipated when conditions such as vomiting, diuretic use, or severe hypertension are present. Thus, a careful and complete history is vital to establishing the etiology. Although the generation and maintenance phases of metabolic alkalosis can be clearly delineated in animal models of metabolic alkalosis, in the clinical setting the genesis of the generation phase may be obscure, even after a careful history. This is especially true if the patient is concealing bulimia or diuretic or laxative abuse.
A physical examination is helpful primarily for assessing the status of body fluid volume. Both deficits and surfeits of volume can and often do accompany metabolic alkalosis depending upon etiology, but they are not causes.
Neurologic symptoms such as apathy, confusion, cardiac arrhythmias, and neuromuscular irritability are observed only when alkalosis is severe (arterial pH >7.55). Although neuromuscular irritability may be readily evident, Chvostek and Trousseau signs are uncommon.
Alkalosis per se has a mild positive inotropic effect on the heart and little or no effect on cardiac rhythm. Cardiac arrhythmias occur primarily because of hypokalemia (see Chapter 4).
Compensatory hypoventilation may contribute to hypoxia or pulmonary infection in very ill or immunocompromised patients.
Many of the clinical features of metabolic alkalosis are dependent largely on the effects of the electrolyte deficits and not on alkalosis per se. K+ depletion may be accompanied by cardiac arrhythmias, nephrogenic diabetes insipidus, and muscle weakness. Similarly, Cl− depletion may be associated with impaired urine concentrating ability, impaired response to loop diuretics, stimulation of renin via a macula densa mechanism, and a reduction in glomerular filtration rate (GFR).
A serum electrolyte profile and an arterial blood gas are necessary to accurately diagnose metabolic alkalosis as outlined above. If the measured Paco2 is within ±3–5 mm Hg of the predicted value, a simple metabolic alkalosis is present. If not, a mixed disorder is present, respiratory acidosis if the Paco2 is higher and respiratory alkalosis if it is lower.
Disequilibrium occurs when generation of HCO3− and resultant elevation of plasma [HCO3−] exceed the capacity of the renal tubule to reabsorb HCO3−. Transient bicarbonaturia with concomitant Na+ or K+ loss ensues until a new steady state is achieved and urinary HCO3− excretion ceases. This ushers in so-called “paradoxic aciduria” sometimes described in stable chronic metabolic alkalosis because the effects of Cl− or K+ depletion prevent urinary excretion of HCO3−.
Additional laboratory tests including serum [Ca2+] and osmolality and urine creatinine, [Cl−], [K+], Na+], osmolality, and Ca2+ for the differential diagnosis are discussed below.
A urine [Cl−] of less than 10 mEq/L characterizes chloride depletion except when a chloruretic diuretic is present in the urine or accompanying profound K+ depletion produces severe tubule dysfunction that induces a Cl− leak. Cl− depletion can be generated by losses from the gut or the kidney; in chloridorrhea, the stool [Cl−] is greater than 90 mEq/L. Fully compensated respiratory acidosis is a Cl− depletion state that will persist after the successful treatment of chronic respiratory acidosis if Cl− repletion has not occurred. In cystic fibrosis (in which renal Cl− handling is normal), high sweat [Cl−] can contribute to Cl− losses with excessive sweating.
When the urine [Cl−] is greater than 20 mEq/L, K+-depletion alkalosis and other miscellaneous disorders are suggested. When K+ depletion is present, the urine K+ excretion is normally less than 20 mEq/day. Thus, in the differential diagnosis of K+-depletion alkalosis, urine K+ excretion greater than 30 mEq/day in the presence of hypokalemia establishes renal K+ wasting and indicates mineralocorticoid excess or an agent that promotes renal K+ wasting. Within this group, K+-depletion alkalosis can be further subdivided by the status of ECF volume and blood pressure. In those diseases characterized by persistent intravascular and ECF volume expansion and consequent hypertension, such as primary aldosteronism or syndromes of apparent mineralocorticoid excess, escape from the Na+-retaining effect of mineralocorticoids occurs but not from the K+-wasting effects thereby promoting alkalosis. The transtubular K+ gradient is a fast and useful test for determining the presence of renal K+ wasting; a value of more than 4 in the setting of hypokalemia is consistent with this abnormality.
In contrast, in Bartter syndrome, Na+ loss as well as both K+ and Cl− losses are associated with normotension. Gitelman’s syndrome is similar but is less severe and is characterized by hypocalciuria not hypercalciuria seen in Bartter’s syndrome. These uncommon syndromes could be confused with some of the commonly concealed causes of metabolic alkalosis such as diuretic or laxative abuse, bulimia, or surreptitious vomiting.
When the urine K+ excretion is less than 20 mEq/day, K+depletion due solely to dietary or gut losses is established. The alkalosis in these disorders is usually mild. More severe alkalosis should suggest additional causative factors, such as concomitant Cl− depletion or base ingestion.
Hypercalcemia, usually in the setting of suppressed parathyroid hormone such as malignancy or vitamin D excess, may uncommonly be associated with alkalosis. In milk-alkali syndrome, multiple factors likely contribute to the alkalosis including vomiting, hypercalcemia, and reduced GFR in addition to the excessive calcium intake.
Hypoalbuminemia may cause mild metabolic alkalosis because of the resultant shift in the buffering capacity of plasma. Patients can develop a metabolic alkalosis if they ingest alkali such as NaHCO3 (eg, baking soda) or calcium carbonate and cannot excrete the excess HCO3− because of renal insufficiency. Obligate excretion of anions with preferential retention of bicarbonate putatively explains the alkalosis that has rarely been associated with some antibiotics.
Transient states of alkalosis are common but usually inconsequential. Intravenous or oral base loading may occur during the transfusion of blood or blood products with a citrate anticoagulant or with the treatment of metabolic acidosis.
Specific treatment is indicated when the arterial pH is greater than 7.55 or the serum bicarbonate concentration is greater than 33 mEq/L. Existing deficits must be replaced and the continued generation of losses should be prevented or blunted to the extent possible.
The Cl− deficit must be replaced with the selection of the accompanying cation based on (1) ECF volume status, (2) the degree of associated K+ depletion, and (3) the degree and reversibility of any depression of GFR. Because the magnitude of the loss of each of these cations is difficult to assess, an empirical clinical approach is recommended. When Cl−and severe K+ depletion coexist, both must be repleted to correct the alkalosis. In patients with overt signs of ECF volume contraction, administration of 3–5 L of 0.15 M NaCl at a minimum is usually necessary to correct volume deficits and metabolic alkalosis. When volume is repleted, further Cl− repletion should be accomplished with KCl unless contraindications are present.
In the clinical setting of ECF volume overload such as congestive cardiac failure in association with Cl− depletion alkalosis, NaCl administration is contraindicated. KCl is the preferred alternative in this setting but it too may be contraindicated or limited because of either concurrent hyperkalemia or renal insufficiency, which could precipitate hyperkalemia.
In addition to volume overload, other serious conditions such as hepatic encephalopathy, cardiac arrhythmias, digitalis cardiotoxicity, or altered mental status also may accompany severe alkalosis. No clinical studies specifically address the impact of the treatment of severe alkalosis on the outcome in these states. However, the high reported mortality of severe alkalosis suggests that to the extent that it may be a contributing factor, alkalosis should be corrected promptly. HCl (0.1 N) administration through a central venous catheter at rates up to 25 mEq/hour should be considered when either Na+ or K+ is not appropriate. The amount of HCl needed to correct alkalosis is calculated by the following formula: 0.5 × body weight (kg) × desired decrement in plasma bicarbonate (mEq/L) with additional amounts to replace any continuing losses of acid. Plasma bicarbonate concentration should be initially restored halfway to normal.
An alternative to HCl is ammonium chloride, which may be administered via a peripheral vein at a rate of not more than 300 mEq NH4+/day; it should be avoided in advanced renal or hepatic insufficiency. The HCl salts of lysine or arginine are available but have been associated with severe hyperkalemia; they are not recommended. Of these agents, HCl is preferred and should be used only as noted.
In the setting of volume overload, acetazolamide 250 mg orally two or three times daily or 5–10 mg/kg intravenously may be effective if GFR is adequate. Because the distal nephron can avidly reabsorb excess Na+ delivery promoted by acetazolamide, the carbonic anhydrase (CA) inhibitors are most effective when used in conjunction with diuretics that have more distal sites of action. Acetazolamide could also be used intermittently to avoid or decrease chloride-depletion matabolic alkalosis (CDA) in edema-forming states such as congestive heart failure treated with loop diuretics. Serum electrolyte composition should be followed serially during its administration since acetazolamide is usually associated with high urinary K+ losses.
CA in erythrocytes and along the pulmonary capillary endothelium participates in the dehydration of bicarbonate to CO2 and its subsequent excretion by the lung. Studies in critically ill patients have shown only minor CO2 retention. Particularly in patients with impaired respiratory function, clinicians should be alert to this potential for CO2 retention with CA inhibition. In such patients, the goal of treatment is the usual stable plasma bicarbonate concentration for that patient and not necessarily a normal concentration.
Other primary or adjunctive therapies should be considered. Villous adenomas require surgical removal. Congenital chloridorrhea does not respond to antidiarrheal agents and dietary repletion of fluid; Cl− and K+ losses are usually required. Omeprazole 20 mg twice daily may substantially decrease diarrhea and obviate the need for dietary electrolyte supplementation; the reduction in the intestinal Cl− load by inhibition of gastric Cl− secretion is presumably the mechanism. When continuing gastric acid losses cannot be prevented, eg, Zollinger–Ellison syndrome, omeprazole or a histamine 2-receptor blocker, eg, cimetidine or ranitidine, will reduce output. These blocking agents have also been used to augment Cl− replacement in patients with unremitting losses due to gastrocystoplasty; on occasion, only surgical revision can correct the alkalosis in these patients.
If renal insufficiency limits the effectiveness of the above therapies or in patients on maintenance dialysis, hemofiltration with replacement infusions of NaCl, exchange of bicarbonate for high bath [Cl−] during hemodialysis, or peritoneal dialysis is an effective means for correcting metabolic alkalosis.
The magnitude of K+ depletion can be estimated from the serum [K+]. The decrease in serum [K+] evoked by alkalosis per se (approximately a 0.5 mEq decrement for each 0.1 increment in arterial pH) accounts for only a modest overestimation of the K+ deficit.
Oral replacement will often suffice unless ileus is present. Oral KCl given in the liquid form diluted with fruit juice or in the slow-release form can be given in doses of up to 40–60 mEq four or five times per day. K+ salts such as citrate, gluconate, or bicarbonate are not appropriate.
If, however, a serious cardiac arrhythmia or generalized paralysis is present, intravenous KCl in concentrations no greater than 60 mEq/L at rates as high as 40 mEq/hour should be preferred. The downregulation of Na+-K+-ATPase in skeletal muscle with K+ depletion states may slow the clearing of the K+ load. Thus, monitoring with electrocardiograms and frequent determinations of serum [K+] are mandatory. Glucose should be omitted from infusions initially because stimulated insulin secretion may cause serum [K+] to decrease even further. However, once repletion is begun, infused glucose will facilitate cellular K+ repletion. If chloruretic diuretics or laxatives are contributing, they should be stopped.
Correction of the K+ deficit reverses the alkalinizing effects of K+ depletion but blockade or removal of the source of mineralocorticoid excess is essential for definitive correction. If the source of aldosterone excess cannot be removed, K+-sparing diuretics will blunt its effects. Amiloride 5–10 mg daily, triamterene 100 mg twice daily, or spironolactone 25–50 mg in single or divided doses daily all are useful. Restriction of Na+ and addition of K+ to the diet also ameliorate the alkalosis and associated hypertension.
In Bartter’s syndrome, the focus of therapy is to prevent urinary K+ loss. Converting enzyme inhibitors have been shown to be effective and are a reasonable first approach. Because of concern for hypotension, low doses should be used initially. Other interventions may also partially correct the alkalosis. The K+-sparing diuretics mentioned above are effective but dietary K+ supplementation is usually also needed. Spironolactone may produce unacceptable gynecomastia in men. Because renal production of prostaglandin E2 is increased and may contribute to Na+, Cl−, and K+ wasting, prostaglandin synthetase inhibitors, such as indomethacin or ibuprofen, may blunt but not completely correct the hypokalemic alkalosis. Since magnesium depletion may contribute to the increase in urinary K+ wasting, hypomagnesemia should be corrected and magnesium stores repleted, if clinically feasible. Oral magnesium oxide in doses of 250–500 mg four times daily (12.5–25 mEq Mg2+) is recommended. However, the degree to which the correction of magnesium depletion blunts the alkalosis is uncertain and magnesium salts often produce an unacceptable degree of diarrhea that can worsen electrolyte imbalance.
Several of the primary disorders of mineralocorticoid excess are treated definitively by tumor ablation or by medication when that cannot be accomplished.
In acute milk-alkali syndrome, cessation of alkali ingestion and the calcium sources (milk, antacids, etc.) and repletion of Cl− and volume usually will lead to the prompt resolution of these abnormalities. The treatment of hypercalcemia is discussed in Chapter 6. For those alkaloses due to alkali loading, cessation of alkali administration and continuation of normal electrolyte intake will usually suffice.
Data on the prevalence and outcome of metabolic alkalosis are sparse. Metabolic alkalosis is common and, when severe, is associated with high morbidity and mortality; in one study, it comprised half of all acid–base disorders. A mortality rate of 45% in patients with a pH of 7.55 and of 80% when the pH was greater than 7.65 has been recorded and confirmed in a separate study (48.5% for alkalemia greater than 7.60). While this relationship between alkalemia and mortality is not necessarily causal, severe alkalosis should be viewed with concern and should be promptly treated.
Metabolic Acidosis
- Reduction in plasma bicarbonate concentration.
- Reduction in blood pH.
- Decreased concentration of carbon dioxide (the compensatory respiratory response).
Metabolic acidosis is one of the four primary acid–base disorders and is caused by a reduction in plasma bicarbonate concentration. It is associated with a reduction in blood pH (termed acidemia) and a decrease in the concentration of carbon dioxide (Pco2), termed the compensatory respiratory response.
Metabolic acidosis can result from several different mechanisms including (1) excessive systemic H+ loads as with ketoacidosis or lactic acidosis, (2) impairment in renal HCO3− generation as with renal failure, (3) extrarenal HCO3− loss as with gastrointestinal HCO3− loss with small bowel diarrhea, and (4) renal HCO3− loss as with proximal renal tubular acidosis (RTA).
The plasma bicarbonate concentration is normally maintained at a constant level of 24–25 mEq/L in males and 22–23 mEq/L in nonpregnant females. Plasma bicarbonate concentration is maintained at these levels, despite ongoing H+ production resulting from metabolism of dietary constituents, because of compensatory equimolar generation of bicarbonate by the kidney (˜70 mmol/day). In addition, since the kidney filters a large quantity of bicarbonate each day, ˜4500 mEq, it must reclaim most of this bicarbonate to maintain a normal plasma bicarbonate concentration.
Approximately 85% of filtered bicarbonate is reclaimed in the proximal tubule. As depicted in Figure 5–1, this bicarbonate is reabsorbed indirectly via the apical sodium–hydrogen exchanger NHE3, and exits the cell via the sodium–bicarbonate cotransporter kNBC1. Membrane-bound carbonic anhydrase IV in the apical and basolateral membranes and cytoplasmic carbonic anhydrase II are necessary for efficient absorption of filtered bicarbonate from the tubular fluid to the systemic circulation.
Figure 5–1.
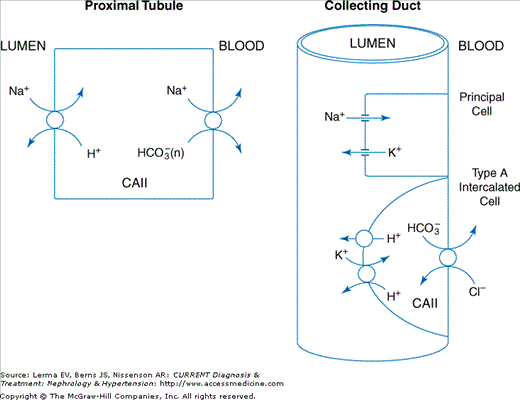
In the proximal tubule luminal HCO3 is absorbed across the apical cell membrane via the Na+/H+ exchanger NHE3. Intracellular HCO3 is transported across the basolateral cell membrane via the electrogenic sodium bicarbonate cotransporter kNBC1 (NBCe1-A). In the collecting duct, the lumen is acidified by Type A intercalated cells that secrete H+ via an H+-ATPase and possible an H+-K+-ATPase. Intracellular HCO3 is transported across the basolateral cell membrane via the anion exchanger AE1. The acidification of the luminal fluid generates H2PO4 (titratable acid) and creates a driving force for the passive diffusion of NH3 into the urine, thereby increasing the urinary excretion of NH4+. Principal cells in the collecting duct are responsible for absorbing Na+ via the epithelial sodium channel (ENaC) and secreting K+ via ROMK. The latter processes are stimulated by aldosterone that binds to receptors on the basolateral membrane of the principal cells.
In addition to absorbing the filtered bicarbonate load, the kidney needs to generate new bicarbonate to match the loss of HCO3 resulting from neutralization of acid generated in the liver from the daily metabolism of dietary protein. The proximal tubule in the kidney generates the new HCO3 primarily from the metabolism of glutamine. In addition to HCO3, NH4+ is also produced. The HCO3 is transported across the basolateral membrane of the proximal tubule cell to the systemic circulation. Were all the NH4+ produced in the proximal tubule transferred along with HCO3 to the systemic circulation, the HCO3 produced in the proximal tubule would be converted to urea in the liver and therefore would be unavailable to buffer the dietary H+ load. As depicted in Figure 5–1, this futile cycle is prevented by intrarenal transport mechanisms that ensure that a portion of the NH4+ is trapped in the lumen of the collecting duct as the result of proton transport by an apically located vacuolar H+-ATPase and possibly H+-K+-ATPase. Luminal proton transport by the H+-ATPase is coupled to basolateral bicarbonate exit via the anion exchanger AE1, with the NH4+ subsequently excreted in the urine.
Collecting duct proton secretion by the H+-ATPase (the primary proton transporter) is modulated by the action of aldosterone, in part by enhancing sodium reabsorption via the epithelial sodium channel (ENaC) to produce a favorable electrical gradient. The kidney generates the remaining new HCO3 by a process called titratable acid (TA) formation/excretion. In this process, secreted H+ (via the proximal tubule NHE3, collecting duct H+-ATPase, and possibly H+-K+-ATPase transporters) bind to HPO42− in the tubule lumen and generates intracellular HCO3 that is transported to the systemic circulation. Clinically, the total effective new bicarbonate generated by the kidney can be quantified by measuring a parameter called net acid excretion, which is equal to the urinary excretion of NH4+ + TA − HCO3−. Under normal acid–base conditions, 40 mEq NH4 and 30 mEq TA are excreted daily while HCO3− excretion is negligible.
The normal response of the body to an H+ load or HCO3− loss involves four processes: (1) Extracellular buffering, (2) intracellular buffering, (3) respiratory compensation, and (4) enhanced renal HCO3− generation. Immediately upon an increase in an acid load, H+ is buffered by plasma HCO3− followed by H+ influx into cells (intracellular buffering). The latter process occurs more slowly, and is completed in 2–4 hours. Approximately 60% of an H+ load is buffered intracellularly, but this can increase dramatically with more severe degrees of metabolic acidosis as bicarbonate buffers are depleted. The degree of intracellular buffering can be indirectly determined by the quantity of bicarbonate required to raise plasma bicarbonate concentration to a certain level (bicarbonate deficit × bicarbonate space). The bicarbonate space is the apparent volume of distribution of administered bicarbonate and is calculated according to the following equation: Bicarbonate space = [0.4 + (2.6/plasma bicarbonate) × lean body weight]. The bicarbonate space can increase from 50% body weight with mild to moderate metabolic acidosis (12–23 mEq/L) to more than 100% body weight with severe metabolic acidosis (plasma bicarbonate concentration <5 mEq/L).
The kidney plays the dominant role in regulating acid–base balance by increasing the quantity of new HCO3− generated. New HCO3− generation increases immediately and achieves a maximal level in approximately 4 days. The quantity of HCO3− generated can increase several fold and is due primarily to enhanced glutamine metabolism.
A fall in plasma bicarbonate concentration and blood pH stimulates receptors in the periphery and central respiratory center increasing alveolar ventilation. The increase in alveolar ventilation creates an inequality between mitochondrial CO2 production and pulmonary CO2 excretion. Pco2 decreases until a new steady state is reached, a process that requires approximately 12–24 hours. The magnitude of the decrease in Pco2 for any given level of sustained metabolic acidosis has been empirically determined and is given by the following relationship: For a given decrease in HCO3 of 10 mEq/L, the Pco2 decreases by approximately 12 mm Hg. When the Pco2 decreases appropriately, the metabolic acidosis is called “compensated” and a simple metabolic acidosis is said to be present. If the Pco2 is above the predicted value, the patient has a coexisting defect in ventilation and a mixed acid–base disturbance, ie, respiratory acidosis and a metabolic acidosis. Conversely, if Paco2 falls below the expected value, the patient has a mixed metabolic acidosis and respiratory alkalosis.
Metabolic acidosis is subdivided into those disorders in which the serum anion gap is normal and those in which it is elevated. The serum anion gap represents the concentration of unmeasured anions minus unmeasured cations: Na+ + K+ + unmeasured cations = Cl− + HCO3− + unmeasured anions. Since the concentration of serum K+ is low, it is not considered in the calculation of the serum anion gap, which is then calculated as Na+ − (Cl− + HCO3). The normal serum anion gap ranges between 8 and 18 mEq/L, with an average of 10–12 mEq/L. However, introduction of a new autoanalyzer method for measuring serum chloride in some clinical laboratories has resulted in a higher value of serum chloride and therefore a lower value for the mean serum anion gap (average of 6–8 mEq/L).
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


