Genetic determinants are thought to play a role. Data from non-alcoholic steatohepatitis (NASH) support this concept. Thus the prevalence of cryptogenic cirrhosis (generally thought to reflect end-stage NASH) is 3.1 fold higher among Hispanic American patients (and 3.9 fold lower in African Americans) compared with the prevalence in Europeans and other Americans. This is despite a similar prevalence of diabetes mellitus, a known risk factor for NASH [2]. Recently, specific single nucleotide polymorphisms (SNPs), which are a stable single base substitution found in more than 1% of the population, have been associated with different rates of fibrosis progression [3,4].
While fibrosis progression rates are useful to predict outcomes in patients with chronic liver disease, they accelerate in HCV as the disease advances [5]. A recent meta analysis examining stage-specific transition probabilities suggested that the probability of transition to a higher stage of fibrosis is greatest between F2 and F3 (4 stage system; Metavir) [6].
Therefore clinicians must make treatment decisions based on host, genetic and environmental factors that may impact on an individual’s risk of disease. In addition, identification of relevant SNPs and gene signatures may not only help tailor therapy for individual patients, but also identify those patients who would benefit most from specific antifibrotic strategies when they become available.
Cellular and Molecular Features of Hepatic Fibrosis (Fig. 6.2)
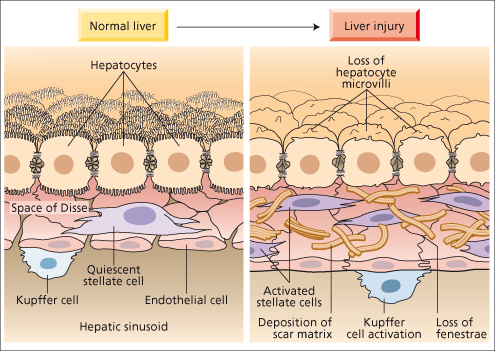
Cellular Anatomy of Sinusoids
Between the sinusoid and hepatocytes, fenestrated endothelial cells line a basement membrane which separates the sinusoidal lumen from the space of Disse. Stellate cells lie in the space of Disse attached to the basement membrane. Kupffer cells adhere to the sinusoidal surface of the fenestrated endothelium. Nutrients and other molecules reach the basal surface of the hepatocyte by passing through the fenestrae of the sinusoidal wall and across the space of Disse. This process is impaired by the cellular and matrix changes seen in liver injury.
Fig. 6.2. Normal cellular and matrix relationship between sinusoid and hepatocyte, and changes after injury.
Extracellular Matrix Composition in Normal Liver and Hepatic Scar tissue
Normal liver has a connective tissue matrix which includes type IV (non-fibrillary) collagen, glycoproteins (including fibronectin and laminin) and proteoglycans (including heparan sulphate). These constituents comprise the low-density basement membrane in the space of Disse, which separates the hepatocytes from the sinusoidal endothelium. This lattice-like matrix provides not only cellular support but also molecular signals that maintain the differentiated functions of cells. The basement membrane allows unimpeded transport of solutes and growth factors between sinusoid and hepatocytes.
After hepatic injury there is a three- to eightfold increase in extracellular matrix, composed predominantly of high-density interstitial fibril-forming collagens (types I and III, rather than type IV) as well as cellular fibronectin, hyaluronic acid and other matrix proteoglycans and glycoconjugates. In addition, there is loss of endothelial cell fenestrations and hepatocyte microvilli associated with this ‘capillarization’ of sinusoids, which impedes the metabolic exchange between blood and liver cells. The gradual accumulation of type I collagen results from both increased synthesis and reduced degradation, and is the hallmark of fibrogenesis.
Stellate Cell Activation: a Central Feature of Hepatic Fibrosis
The hepatic stellate cell (HSC) (also called lipocyte, fat-storing cell, Ito cell, pericyte) is the principal cell involved in fibrogenesis. It lies within the space of Disse and in direct contact with hepatocytes, endothelial cells, inflammatory cells and nerve fibres (Fig. 6.2). In the normal liver, these cells have intracellular droplets containing vitamin A. They contain 40–70% of the body stores of retinoids.
In its quiescent state the HSC produces predominantly type IV collagen, the collagen characteristic of a normal basement membrane. With injury, it undergoes phenotypic changes referred to as ‘activation’, characterized by loss of retinoid droplets, cellular proliferation, increased endoplasmic reticulum, increased contractility with expression of smooth muscle specific α-actin, and secretion of cytokines/ chemokines (Fig. 6.3). This phenotypic switch is also characterized by production of type I collagen, the high-density interstitial collagen characteristic of the cirrhotic liver, as well as matrix-degrading enzymes.
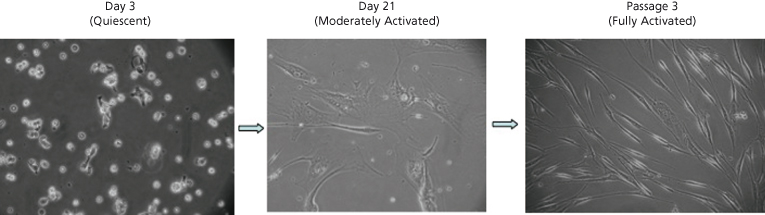
Fig. 6.3. Morphological changes in human hepatic stellate cells (HSCs) during culture-induced activation [11]. HSCs are isolated by density centrifugation from normal liver. Plated on plastic they are initially vitamin A-rich cells, exhibiting autofluorescence. Subsequently, they lose their vitamin A droplets, becoming more proliferative and spindle shaped. Culture-induced activation is a model system used to study in vivo activation. (Phase-contrast microscopy, ×200.)
Stellate cell activation is a central event in hepatic fibrosis and can be conceptualized as occurring in at least two stages: (1) initiation and (2) perpetuation (Fig. 6.4).
Fig. 6.4. Pathways of stellate cell (HSC) activation. Stellate cell activation can be divided into two phases: initiation and activation. Initiation is provoked by soluble stimuli that include oxidant stress signals (reactive oxygen intermediates), apoptotic bodies, lipopolysaccharide (LPS) and paracrine stimuli from neighbouring cell types including hepatic macrophages (Kupffer cells), sinusoidal endothelium and hepatocytes. Perpetuation follows, characterized by a number of specific phenotypic changes including proliferation, contractility, fibrogenesis, altered matrix degradation, chemotaxis, and inflammatory signalling. PDGF, platelet derived growth factor; VEGF, vascular endothelial growth factor; FGF, fibroblast growth factor; ET-1, endothelin-1; NO, nitric oxide; TGFβ1, transforming growth factor-β1; CTGF, connective tissue growth factor; MMP, matrix metalloproteinase; MT-MMP, membrane type matrix metalloproteinase; TRAIL, TNF-related apoptosis-inducing ligand;TIMP, tissue inhibitor of metalloproteinase; TLR, toll like receptor.
Modified from [7], with permission.
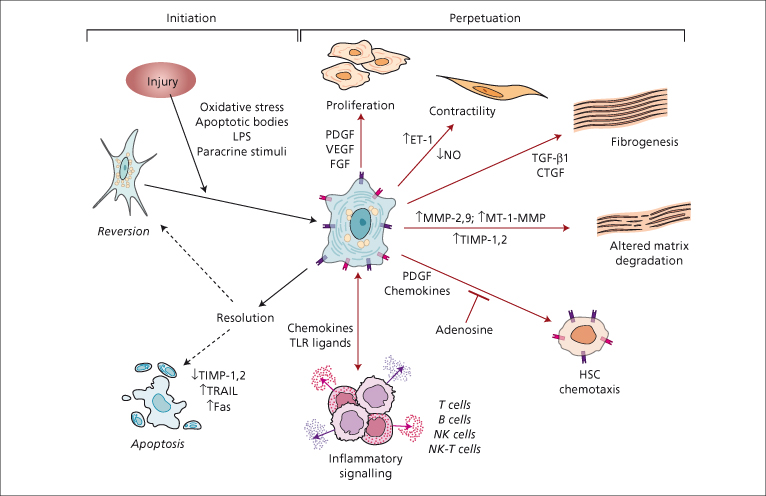
Initiation refers to early events, including rapid changes in gene expression and a cellular phenotype that renders HSCs responsive to cytokines and other stimuli. Initiation is provoked by different factors depending on disease aetiology. Stimuli include oxidant stress signals (reactive oxygen intermediates), apoptotic bodies and lipopolysaccharide. Moreover, the rapid, disruptive effects of liver injury result in early changes in the extracellular matrix (ECM) composition and alter the homeostasis of neighbouring cells such as hepatic macrophages (Kupffer cells), sinusoidal endothelium and hepatocytes, resulting in paracrine stimuli that ‘prime’ the HSC to respond to a host of growth factors and cytokines.
Perpetuation involves cellular events that amplify the activated phenotype through enhanced cytokine expression and responsiveness, and the acquisition of features critical to the development of fibrosis.
These signals provide the impetus for scar formation through:
- enhanced HSC proliferation, contractility and fibrogenesis;
- altered matrix degradation;
- HSC chemotaxis;
- direct interactions between HSCs and the immune system;
- secretion of proinflammatory mediators.
Once the initiating injury signal is eliminated (i.e. treatment of underlying disease, discontinuation of hepatotoxins such as ethanol), HSCs either revert to the quiescent phenotype or are removed from the liver through programmed cell death, or apoptosis. This paradigm has provided the framework for the development of numerous antifibrotic approaches (see below).
Proliferation
With activation, HSCs proliferate rapidly. Platelet derived growth factor (PDGF-β) is the most potent mitogenic factor for HSCs by acting through its receptor, β-PDGFR [7]. Both the PDGF ligand and receptor are rapidly induced in vivo and in culture as HSCs activate [8,9]. Other stellate cell mitogens include vascular endothelial growth factor (VEGF), thrombin, endothelial growth factor (EGF), transforming growth factor-α (TGF-α), keratinocyte growth factor, fibroblast growth factor (FGF), insulin-like growth factor IGF-1 and CXCL12 [10,11].
Contractility
During liver injury, the normally quiescent HSC also acquires ‘myogenic’ features including expression of alpha smooth muscle actin [12] and myosin [13], which confer contractile properties. Given their location within the space of Disse, HSC contractility contributes to increased portal resistance even with early fibrosis. This may be more reversible than when portal pressure is increased as a result of thickened septae and lobular distortion characteristic of advanced fibrosis. Endothelin-1 and nitric oxide are key regulators that control HSC contractility through their mutually antagonistic activities. Contractility is also effected by many other factors including angiotensin II, eicosanoids, atrial naturetic peptide, somatostatin and carbon monoxide, among others [14,15].
Fibrogenesis
The production of type I collagen is the cardinal feature of the activated stellate cell. While other cytokines are important for the induction of HSC-derived collagen I, TGF-β1 remains the most potent fibrogenic cytokine. Cellular sources of TGF-β1 in chronic liver injury include sinusoidal endothelial cells, Kupffer cells and HSCs. Therefore, both autocrine and paracrine loops contribute to the development of liver fibrosis [16]. Other profibrogenic cytokines include CTGF, FGF and VEGF. Angiotensin II, the main effector of the renin–angiotensin system, is a functional cytokine that is a potential activator of collagen production in HSCs and a target of antifibrotic therapies [17].
Chemotaxis
Since fibrosis is a normal wound-healing response to encapsulate injury, it is not surprising that the HSCs migrate towards sites of injury driven by chemoattractants, which include PDGF [18], monocyte chemotactic protein-1 (MCP-1) [19] and CXCR3 ligands [20]. This ability to migrate to sites of injury may also be important for interactions with the immune system.
Inflammatory Signalling
HSCs are also effectors in the liver’s immune response to injury. HSCs secrete proinflammatory cytokines/ chemokines such as MCP-1, underscoring their ability to promote inflammation rather than simply serving as a passive target of inflammatory cytokines. HSCs, like dendritic cells, can also function as professional antigen presenting cells by efficiently presenting antigen to MHC-I and MHC-II-restricted T cells in vitro and stimulating lymphocyte proliferation [21,22]. In addition, signalling by TLR4 (toll-like receptor 4) in HSCs in response to bacterial lipopolysaccharide further implicates this cell type in the liver’s innate immune response to injury [23]. Interestingly, specific TLR4 SNPs contribute to fibrosis progression in HCV infection, providing a direct link between genetic risk and disease pathogenesis [4].
Other Collagen-Producing Cells
While stellate cell activation is clearly central to most fibrosing chronic liver injury, other collagen-producing cells may also contribute to ECM accumulation in the liver. The relative contribution to fibrogenesis by different cell types may vary according to the aetiology of the liver disease. Within the liver, portal myofibroblasts are particularly important in biliary fibrosis [24]. Conversion, or ‘transdifferentiation’, of epithelial cells of the liver, hepatocytes and biliary epithelial cells, to become mesenchymal cells, a process referred to as epithelial–mesencyhmal transition (EMT), has been demonstrated in animal models but the contribution of EMT to human liver disease is not clear [25].
Liver sinusoidal endothelial cells also make collagen I. However these cells are particularly important in initiating the fibrogenic process through production of a splice variant of cellular fibronectin, called fibronectin extracellular domain A. Fibronectin extracellular domain A is produced early in animal models, and along with TGF-β1 contributes to myofibroblast differentiation [26].
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


