An alternative classification groups liver failure into fulminant, subfulminant and late-onset hepatic failure. Fulminant hepatic failure is when time from jaundice to encephalopathy is less than 2 weeks and subfulminant hepatic failure when time is greater than 2 weeks [7]. Late-onset liver failure describes encephalopathy developing more than 8 weeks (but less than 24 weeks) after the first symptoms [8].
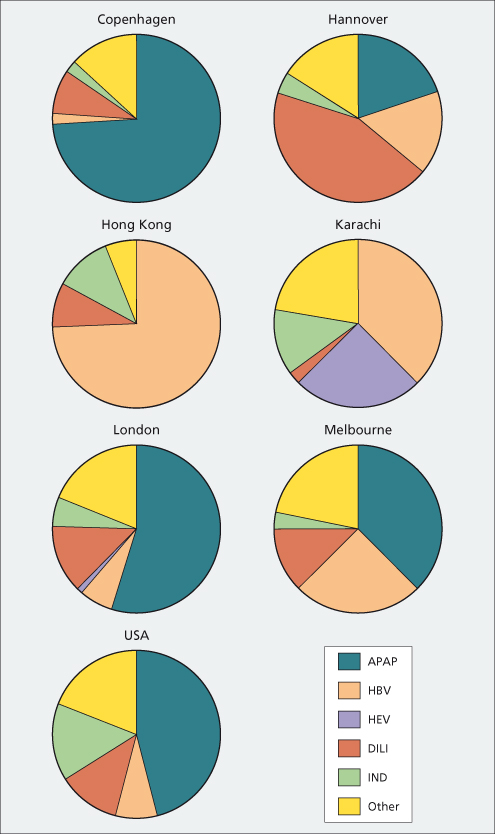
Epidemiology and Aetiologies (Fig. 5.1, Table 5.2)
The most common cause of acute liver failure worldwide is viral hepatitis. Asia and the developing world contribute the most cases, probably because of higher prevalence of viral hepatitis and less exposure to drugs. Most Western countries have shown a decline in viral aetiologies during the last decade and an increase in paracetamol and idiosyncratic drug-induced cases of liver failure [9]. The exception to this pattern is Spain, where paracetamol is not readily available and hepatitis B remains the most common identifiable cause [10]. In the USA and UK, paracetamol self-poisoning accounts for 45–60% of acute liver failure with idiosyncratic drug reactions accounting for 12% of cases [11].
Table 5.2. Causes of acute liver failure
| Infections |
| Hepatitis A, B, C, D, E |
| Herpes simplex |
| Epstein–Barr virus |
| Cytomegalovirus |
| Transfusion-transmitted virus (TTV) |
| Dengue fever |
| Drugs and toxins |
| Paracetamol |
| Carbon tetrachloride |
| Idiosyncratic drug reactions* |
| Mushroom poisoning |
| Sea anemone sting |
| Ischaemic |
| Cardiogenic shock |
| Hypotension |
| Heat stroke |
| Cocaine, methamphetamines, ephedrine |
| Vascular |
| Acute Budd–Chiari syndrome |
| Sinusoidal obstruction syndrome |
| Miscellaneous |
| Wilson’s disease |
| Acute fatty liver of pregnancy |
| Eclampsia/ HELLP syndrome |
| Malignancy |
| Primary graft non-function after liver transplantation |
Viral Hepatitis
The hepatitis virus responsible varies from one geographical location to another. In the USA, only 11% of acute liver failure is viral, with 2.5% being from hepatitis A, 7.7% from hepatitis B and the remainder from other viruses [12]. In India, over 95% of acute liver failure is due to viruses, with 40% secondary to hepatitis E and 25–30% to hepatitis B [13]. Even with the development of the hepatitis B vaccine, over half of acute liver failure cases in Greece are due to hepatitis B, though this has decreased from over 70% in the 1980s [14]. Hepatitis B accounts for almost 80% of acute liver failure cases and represents two-thirds of liver transplants [15]. Although these cases are designated as acute, many probably represent flares of chronic hepatitis B, that arises from changes in balance between immune response and virus replication.
Although hepatitis A has a broad distribution, it remains a rare cause of acute liver failure and is declining rapidly in the USA. Indicators of poor prognosis include creatinine above 2.0 mg/dL, alanine aminotransferase less than 2600 IU/mL, and need for ventilatory or pressor support [16]. Those with chronic liver disease may be more susceptible, with liver failure occurring in up to 41% of patients with underlying chronic hepatitis C who contract hepatitis A [17]. Vaccination against hepatitis A as well as hepatitis B is recommended for those with chronic liver disease due to hepatitis C as well as other causes [18].
Liver failure occurs in approximately 1% of patients with acute hepatitis B and jaundice [19]. IgM hepatitis B core antibody (anti-HBc) will be positive in fulminant hepatitis B. However, diagnosis may be obscured since one-third to one-half of patients become seronegative for hepatitis B surface antigen (HBsAg) after a few days [20,21]. Early studies suggested that core promoter and precore variants may increase the risk of acute liver failure but this has not been validated in later studies [22–24]. Superinfection with hepatitis D virus precipitates 4% of acute liver failure in hepatitis B endemic areas but is uncommon in the USA [25]. Reactivation of viral replication in inactive carriers of hepatitis B can trigger acute liver failure, either spontaneously or due to chemotherapy or immunosuppression for organ transplantation [26].
Hepatitis C as a cause of acute liver failure appears to be extremely rare in the USA and Europe but may be more common in the East. In some series, HCV RNA was detected in up to half of patients with non-A, non-B fulminant hepatitis. However, markers of chronic hepatitis B infection can be suppressed by acute HCV infection, resulting in erroneous attribution of the liver damage to hepatitis C alone [15].
Hepatitis E, like hepatitis A, is spread primarily through contaminated water supplies. While rare in the West, it is a leading cause of acute liver failure in endemic areas such as India, Central and Southeast Asia, Mexico and North Africa [13]. Pregnant women are particularly at risk for acute liver failure but pregnancy itself does not appear to impart a worse prognosis once liver failure is present [27].
Other viruses can cause a fatal hepatic necrosis especially in immunocompromised individuals. These include herpes simplex, varicella zoster, cytomegalovirus, adenoviruses, Epstein–Barr, dengue fever and parvovirus B19 [28–32]. In a survey of indeterminate acute liver failure patients in the US, no instance of hepatitis E or parvovirus B19 was identified [33].
Paracetamol
Paracetamol is a dose-related toxin, the most common suicidal agent in the UK (Chapter 24). In 1998, legislation enacted in the UK mandated that paracetamol be sold in blister packs with a limit on the number of tablets obtainable without prescription. Since then, there has been a reduction in the frequency of severe paracetamol-related hepatotoxicity, the number listed for transplant and deaths [34]. However, in the USA paracetamol remains the leading cause of acute liver failure, comprising 46% of cases in the US Acute Liver Failure Study Group (ALFSG) database. In contrast to the UK, nearly half of the paracetamol acute liver failure cases were considered unintentional and strongly related to over use and abuse of narcotic-containing compounds [35].
The characteristic picture of paracetamol toxicity includes very high serum aspartate or alanine aminotransferase levels (reported up to 48 000 IU/L) accompanied by relatively low bilirubin levels (4–6 mg/dL), exemplifying the acuity of the injury [36]. Patients in the USA with acetaminophen-related acute liver failure are equally divided between those who are suicidal and those who are considered to be unintentional overdoses. The unintentional patient typically will have taken medication above the daily recommended dose for several days for a specific cause of pain, denies suicidal intent and fails to recognize the risk of toxicity [37]. Alcohol may be an important cofactor in the non-suicidal patient. Among patients who developed liver failure and reported taking < 4 gm/day, alcohol abuse was present in 65% [35].
Other Aetiologies
Idiosyncratic drug reactions may cause acute liver failure in up to 12% of cases in Western countries and much fewer in developing nations (Table 5.3). Cases are typically subacute with moderately elevated aminotransferases and high bilirubin levels and poor survival without transplantation (approximately 25% in most series) [38]. The most frequent culprits are antituberculosis medications, non-steroidal anti-inflammatory drugs, anaesthetic agents and antiseizure medications [39]. Acute liver failure is also reported with the recreational drug ‘ecstasy’ (3,4-methylene dioxymetamphetamine) [40]. Herbal remedies, especially those containing green tea extract, have been implicated in acute liver failure [41].
Table 5.3. Some drugs that may cause idiosyncratic liver failure
| Injury leading to acute liver failure [38]: | |
| Isoniazid | Isoflurane |
| Sulfonamides | Lisinopril |
| Phenytoin | Nicotinic acid |
| Nitrofurantoin | Imipramine |
| Propylthiouracil | Gemtuzumab |
| Halothane | Amphetamines/ ecstasy |
| Disulfiram | Labetalol |
| Valproic acid | Etoposide |
| Amiodarone | Flutamide |
| Dapsone | Tolcapone |
| Herbals* | Quetiapine |
| Didanosine | Nefazodone |
| Efavirenz | Allopurinol |
| Metformin | Methyldopa |
| Ofloxacin | Ketoconazole |
| Ciproflixacin | |
| Pyrazinamide | |
| Troglitazone | |
| Diclofenac | |
| Combination agents with enhanced toxicity: | |
| Trimethoprim–sulfamethoxazole | |
| Rifampin–isoniazid | |
| Amoxicillin–clavulanate | |
* Some herbal products/ dietary supplements that have been associated with hepatotoxicity include: kava kava, chaparral, skullcap, germander, pennyroyal, jin bu huan, heliotrope, rattleweed, comfrey, sunnhemp, senecio, impila, greater celandine, gum thistle, he shon wu, ma huang, lipokinetix, bai-fang herbs, Hydroxycut®.
Mushroom poisoning, usually Amanita phalloides, can cause acute liver failure; it is preceded by muscarinic effects, such as profuse sweating, vomiting and diarrhoea within hours to a day of ingestion. Mortality approaches 30%. Early recognition is important to optimize supportive measures and to be alerted to the possibility of liver failure [42].
Pregnant women may develop hepatic necrosis during the third trimester, due to eclampsia and/or fatty liver (Chapter 30).
Vascular causes of ischaemic hepatitis include low cardiac output in a patient with underlying cardiac disease, systemic hypotension as seen with sepsis or cardiac events, acute Budd–Chiari syndrome, and sinusoidal-obstruction syndrome occurring after bone marrow transplantation. ‘Shock liver’ is rarely fatal and prognosis depends on the patient’s underlying medical condition, whereas Budd–Chiari and sinusoidal-obstruction syndrome have poorer outcomes [43,44].
Massive infiltration of the liver with tumour such as in lymphoma can lead to acute liver failure. Such a cause should be considered in the differential diagnosis since liver transplantation is contraindicated, and specific therapy may be life saving [45].
Wilson’s disease may present with liver failure associated with profound haemolytic anaemia and renal failure. Patients are usually diagnosed between the ages of 5 and 40 years. Fulminant hepatic failure due to Wilson’s disease is universally fatal without liver transplantation [46].
Autoimmune hepatitis may rarely present as acute liver failure and may be the underlying cause in patients with indeterminate disease [47].
Clinical Features
The patient with acute liver failure typically develops non-specific symptoms such as nausea, vomiting, and malaise, jaundice and signs of hepatic encephalopathy, evolving relatively quickly. The liver is often shrunken due to loss of hepatic mass and may be as small as 600 g in size (normal approximately 1600 g). Declining hepatocellular function impairs synthesis of clotting factors and glucose leading to coagulopathy and hypoglycaemia. Metabolic acidosis results from reduced clearance and increased production of lactate. Tachycardia, hypotension, hyperventilation and fever may occur and signs of the systemic inflammatory response may be present. Transfer of the patient to a specialist liver centre with a transplantation service should be done earlier rather than later. The presence of any degree of encephalopathy necessitates an immediate transfer to a transplant facility unless contraindications are present.
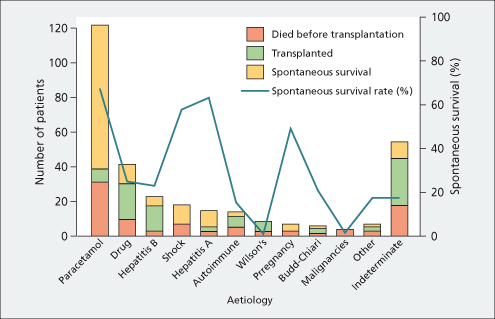
Patients with a more gradual onset of hepatic insufficiency (over weeks rather than days, and variously called subfulminant, subacute or late onset) infrequently develop cerebral oedema. Ascites, oedema and renal failure are more likely in this slowly evolving setting; outcome depends on the underlying aetiology (Fig. 5.2) [3]. Those patients that survive without transplant usually have a complete recovery [48].
Distinction from Chronic Liver Disease
A note should be made of any history of liver disease, the duration of symptoms and the presence of a hard liver, marked splenomegaly and vascular spiders on the skin. In patients with evidence of chronic liver disease, perform a full evaluation for potential causes of decompensation. Infection, gastrointestinal bleeding, dehydration, sedatives and alcohol are common culprits. In the alcoholic, recent heavy drinking can add acute hepatitis to underlying chronic liver disease. In these circumstances the liver is large. Acute alcoholic hepatitis is potentially reversible and merits aggressive supportive effort.
Initial Investigations
Laboratory (Table 5.4)
A broad array of blood tests must be ordered initially to establish the aetiology and severity of the injury. Assessing prognosis is central to management. Despite some drawbacks, the prothrombin time/ international normalized ratio (INR) reflects the liver’s synthetic function and is central to the assessment of the severity, absent plasma replacement. Elevated white blood cell count may signify an underlying infection. Low haemoglobin level may be a sign of haemolysis, often seen in Wilson’s disease [49] or indicate gastrointestinal blood loss. The platelet count is low in nearly 80% for unclear reasons [3].
Table 5.4. Investigations of acute liver failure
| Haematology |
| Complete blood count: white blood cells, haemoglobin, haematocrit, anti-hepatitis B core |
| Coagulation panel: prothrombin time/INR, factor V |
| Blood group |
| Biochemical |
| Serum chemistries: sodium, potassium, bicarbonate, chloride, urea, creatinine, calcium, magnesium, phosphate, glucose |
| Hepatic panel: aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase, albumin, total protein, total bilirubin |
| Arterial blood gas |
| pH, PaCO2, PaO2, lactate, ammonia |
| Virology |
| Hepatitis B surface antigen and IgM anticore |
| Hepatitis A (IgM) antibody |
| Hepatitis C antibody (for underlying chronic infection) |
| HCV RNA |
| IgM hepatitis E antibody (in endemic areas) |
| Hepatitis D antibody if hepatitis B positive |
| HSV, CMV, EBV PCR (if history of immunosuppression) |
| Human immunodeficiency virus (if considering transplantation) |
| Autoimmune markers |
| Antinuclear antibody (ANA) |
| Antismooth-muscle antibody (SMA) |
| Antiliver/ kidney microsome 1 (ALKM1) |
| Immunoglobulins |
| Toxicology |
| Paracetamol level |
| Blood alcohol |
| Urine drug screen |
| Miscellaneous |
| Urine copper |
| Pregnancy test |
| Microbiology |
| Blood culture, aerobic and anaerobic |
| Urine culture and microscopy |
| Sputum culture and microscopy |
| Other studies |
| Chest X-ray, electrocardiogram |
| Liver ultrasound with Dopplers |
| Electroencephalogram (EEG), non-contrast head CT (in some cases) |
| Liver biopsy |
Serum chemistries including glucose, electrolytes, urea and creatinine are measured. Hypoglycaemia can be severe, contributing to altered mental status. Sodium, potassium and phosphorus are commonly low as is carbon dioxide in the setting of hyperventilation. Liver function tests including bilirubin, aminotransferases, alkaline phosphatase, total protein and albumin are routinely done and are usually markedly abnormal. The aminotransferases have little prognostic value since levels can fall as the patient’s condition worsens or improves. Acidosis is common in paracetamol-related liver failure and is a sign of poor prognosis.
Viral serologies may identify potential aetiologies; the appropriate IgM antibodies are useful in identifying hepatitis A and B. HBsAg may have been cleared but hepatitis B surface antibody (anti-HBs) will generally not be present. Serum HBV DNA usually falls quickly and may be undetectable. In those positive for HBV, serum hepatitis D antibody should be sought. Antihepatitis C virus antibody (anti-HCV) and PCR for HCV RNA are required for diagnosis of HCV-related acute hepatic failure (Chapter 20). Hepatitis E serology IgM should be done if other aetiologies are excluded since HEV can be seen in non-endemic areas and in persons who have not travelled.
Herpes simplex virus and varicella zoster virus serologies and PCR should be checked, especially in immunosuppressed or pregnant patients, as these require specific treatment [50,51]. Liver biopsy, if performed, will show specific viral inclusions, but the presence of skin lesions merit initiation of acyclovir therapy.
Paracetamol level and toxicology screen should be obtained. However, paracetamol levels are often undetectable in the setting of unintentional cases if symptoms have already developed. Hepatotoxicity cannot be accurately predicted using the standard Rumack–Matthew nomogram if the precise time of ingestion is unknown, if the patient took multiple overdoses over time or if the patient had been taking extended-release preparations [52–55]. Paracetamol levels may be falsely elevated with bilirubin concentrations more than 10 mg/dL depending on the assay [56]. Markedly elevated aminotransferases, often more than 3500 IU/L, are strongly suggestive of paracetamol toxicity [36]. Paracetamol protein adducts correlate with hepatotoxicity and remain detectable for up to 12 days after ingestion. The detection of adducts, currently a research-only assay, may provide an important clinical tool when it becomes more widely available [57].
Antinuclear antibodies, smooth muscle antibodies, antibodies to liver/ kidney microsomes type 1 and immunoglobulin levels should be checked for possible autoimmune hepatitis; liver biopsy may help to establish the diagnosis and is encouraged in indeterminate case settings [58].
Serum ceruloplasmin is unhelpful in fulminant Wilson’s disease since levels are low in nearly 50% of all forms of acute liver failure [59]. Measurement of the ratio of alkaline phosphatase to bilirubin of less than 4 and aspartate aminotransferase to alanine aminotransferase greater than 2.2 is highly accurate in diagnosing Wilson’s disease and can be obtained more rapidly than tests such as urine copper [60].
Electroencephalogram (EEG)
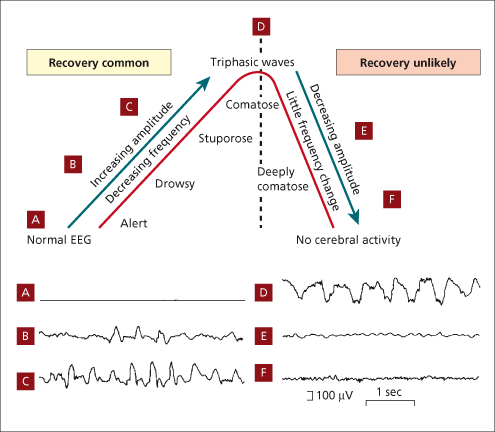
Continuous EEG recording shows slowing of cortical activity and up to 50% of patients with acute liver failure to have subclinical seizure/ epileptiform activity (Fig. 5.3). This is not recognized clinically without EEG because the patient is usually paralysed and ventilated. EEG monitoring is controversial since prophylactic phenytoin is of unproven value [61,62].
Fig. 5.3. Evolution of the EEG in liver failure. The progression from grade A to D is marked by increasing amplitude, decreasing frequency and increasing drowsiness. At D, triphasic waves appear and the interrupted line indicates the limit beyond which recovery is unlikely. From E to F amplitude decreases with little frequency change and at F there is no cerebral activity.
Computed Tomography (CT)
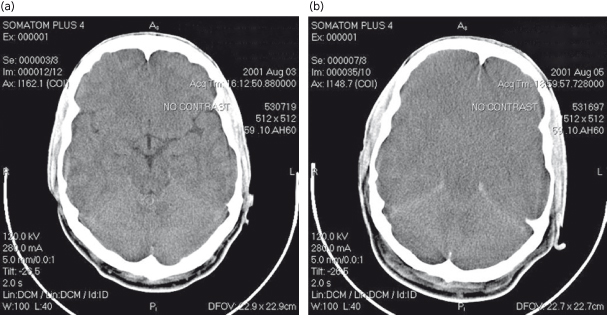
Non-contrast computed tomography of the brain is insensitive for detecting intracranial hypertension (Fig. 5.4) but may help rule out other pathology, such as haemorrhage. The yield of such studies is low and may not justify the risk of moving a critically ill patient [63–65].
Fig. 5.4. Cerebral oedema on CT scanning in a patient with acute liver failure. (a) Head CT at presentation showing clear demarcation between white and gray matter. (b) Head CT 48 h later demonstrating loss of demarcation between white and gray matter and effacement of sulci.
Abdominal Imaging and Liver Biopsy
Abdominal ultrasound is used to assess for vasculature patency and mass lesions. Hepatic nodularity is commonly seen in the acute setting, reflecting regenerative nodules rather than cirrhosis [66]. CT scanning will show a reduction in liver size but correlation of liver size with survival is imprecise.
Liver histology can show considerable variability of necrosis that may be prognostically misleading [67]. Sampling error precludes use of liver biopsy for prognosis. Transjugular liver biopsy should be performed if there is any suspicion of malignancy or autoimmune hepatitis [38]. It may aid in diagnosis (Fig. 5.5). In a retrospective study a liver volume of less than 1000 mL and/or hepatic parenchymal necrosis of greater than 50% indicated a poor prognosis, but findings above these two thresholds did not necessarily indicate a good outcome [68].
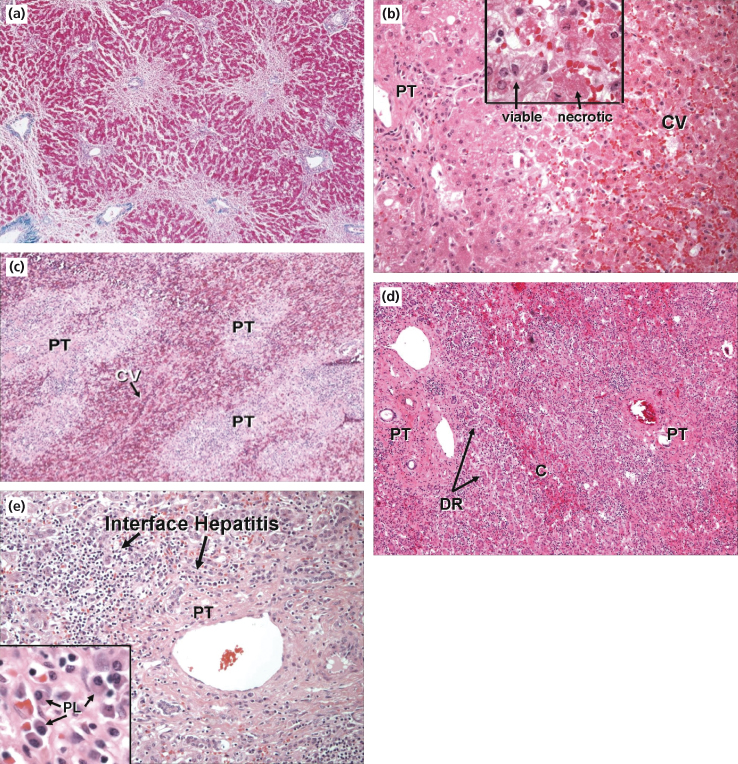
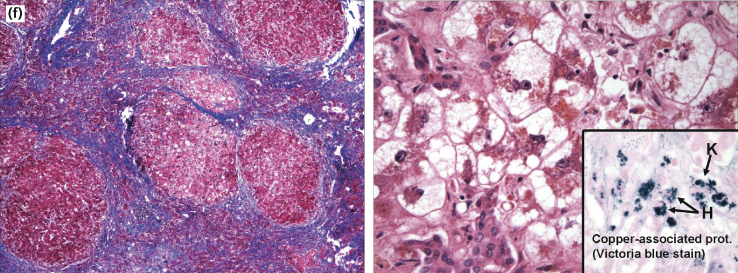
Fig. 5.5. Histological features of acute liver failure. (a) Autopsy specimen from a patient who died of cerebral oedema following paracetamol ingestion. Haematoxylin and eosin slide demonstrates centrilobular necrosis with surviving periportal hepatocytes. (b) Explanted liver in a case of paracetamol toxicity. Necrotic hepatocytes with eosinophilic cytoplasm around the central vein (CV) with viable hepatocytes surrounding the portal tracts (PT). (c) Explanted liver of a patient with fulminant acute hepatitis B. Multilobular hepatic necrosis centred around the central vein (CV) with inflammatory infiltration of the portal tracts (PT). (d) Explanted liver of a patient with acute liver failure following black cohosh ingestion. Massive hepatic necrosis with minimal centrilobular (C) parenchyma remaining. Portal tracts (PT) expanded by inflammatory cells and bile duct reaction (DR). (e) Higher magnification demonstrating features of autoimmune hepatitis with interface hepatitis and multiple plasma cells (PL). (f) Explanted liver of a 20-year-old patient with Wilson’s disease. Trichrome stain shows cirrhosis with bands of fibrosis surrounding regenerative nodules. Haematoxylin and eosin slide (right) shows severe hepatocyte injury with ballooning degeneration, microsteatosis and cholestasis. Victoria blue stain highlights copper pigment within the hepatocytes (H) and Kupffer cells (K).
Histology courtesy of Jay Lefkowitch, MD, Columbia University College of Physicians and Surgeons, New York, NY.
Complications and Management of Acute Liver Failure
Acute liver failure represents a syndrome precipitated by various causes rather than a single disease. In addition to the defining hepatic encephalopathy and coagulopathy, significant hepatocyte death incites a cytokine storm which may result in systemic inflammatory response syndrome (SIRS), multiple system organ failure and ultimately death [69,70]. Treatment has focused on the management of complications except for a few aetiology specific therapies (Table 5.5). However, a recent double-blind placebo controlled trial of intravenous N-acetylcysteine (NAC) demonstrated improved transplant-free survival in non-paracetamol acute liver failure patients with early stage encephalopathy, perhaps offering a common therapy for fulminant hepatic failure [71].
Table 5.5. Intensive care of acute liver failure
| Cerebral oedema/ intracranial hypertension |
| Grade 1/2 encephalopathy |
| Consider transfer to liver transplant facility and listing for transplantation |
| Head CT: rule out other causes of decreased mental status; little utility to identify cerebral oedema |
| Avoid stimulation, avoid sedation if possible |
| Antibiotics: surveillance and treatment of infection required; prophylaxis possibly helpful for unexplained deterioration |
| Lactulose: possibly helpful |
| Grade 3/4 encephalopathy |
| Continue management strategies listed above |
| Intubate trachea (may require sedation) |
| Elevate head of bed |
| Consider placement of ICP monitoring device |
| Immediate treatment of seizures required; prophylaxis of unclear value |
| Mannitol (0.25–1 g/kg i.v. bolus): use for severe elevation of ICP or first clinical signs of herniation, monitor urine output and serum osmolarity |
| Hypertonic saline (30% 5–10 mL/h): goal serum sodium 145–155 mmol/L, avoid rapid correction |
| Hyperventilation: effects short-lived; may use for impending herniation |
| Barbiturate coma (pentobarbital 3–5 mg/kg i.v. bolus then 1–3 mg/kg/h or thiopental 5–10 mg/kg i.v. bolus then 3–5 mg/kg/h): watch for hypotension |
| Continuous renal replacement, hypothermia (32–35°C), i.v. indomethacin (25 mg bolus) may have some benefit in very refractory cases |
| Infection |
| Aseptic techniques |
| Surveillance for and prompt antimicrobial treatment of infection required |
| Antifungal coverage for patients not responding to broad spectrum antibiotics |
| Antibiotic prophylaxis possibly helpful but not proven |
| Coagulopathy |
| Vitamin K (10 mg i.v. or subcutaneous) |
| FFP: give only for invasive procedures or active bleeding |
| Platelets: give for platelet counts <10 000/mm3 or invasive procedures |
| Recombinant activated factor VII (40 µg/kg bolus): give 30 min before procedure if coagulopathy refractory to FFP |
| Cryoprecipitate: for fibrinogen <100 mg/dL and bleeding |
| Exchange plasmapheresis: allows transfusion of large amount of FFP |
| Prophylaxis for stress ulceration: give H2 blocker or PPI |
| Haemodynamics/ renal failure |
| Arterial line |
| Pulmonary artery catheterization |
| Volume replacement |
| Check cortisol and cosyntropin stimulation test; hydrocortisone 200–300 mg/day for adrenal insufficiency |
| Pressor support (noradrenaline preferred over dopamine or adrenaline) as needed to maintain adequate mean arterial pressure |
| Avoid nephrotoxic agents |
| Continuous modes of haemodialysis if needed, such as venovenous haemodialysis |
| Vasopressin: potentially harmful |
| Pulmonary |
| Sedation for endotracheal intubation and suctioning to prevent increased ICP |
| Ventilator management: tidal volumes 6 mL/kg, low PEEP, aim for PaCO2 30–40 mmHg |
| Metabolic concerns |
| Follow closely: glucose, potassium, magnesium, phosphate |
| Consider nutrition: enteral feedings preferred over total parenteral nutrition |
ICP, intracranial pressure; FFP, fresh frozen plasma; PEEP, positive end-expiratory pressure.
Hepatic Encephalopathy
Hepatic encephalopathy and cerebral oedema with raised intracranial pressure (ICP) are hallmarks of acute liver failure (Fig. 5.6). Once stupor develops with or without decerebrate posturing (stage 3–4 encephalopathy), cerebral oedema is likely.
Fig. 5.6. Brain dysfunction in acute liver failure. Proposed interrelation of metabolic encephalopathy, intracranial pressure and changes in cerebral blood flow during the progression of the disease. HE, hepatic encephalopathy [72].
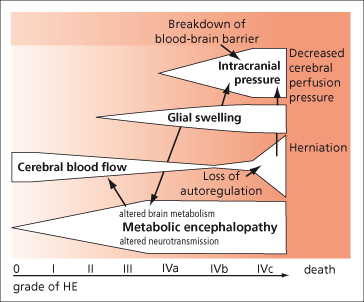
The pathogenesis of hepatic encephalopathy is multifactorial (Chapter 8) and centres on failure of the liver to remove toxic, mainly gut-derived, substances from the circulation. Arterial ammonia levels rise and appear to contribute to astrocyte swelling. Levels greater than 150 to 200 mmol/L have been shown to correlate with cerebral oedema and herniation [73–75]. In contrast to the coma of cirrhotic patients, portal–systemic encephalopathy due to shunting of blood past the liver is of minor importance.
The onset of encephalopathy is often sudden, may precede jaundice, and, unlike chronic liver disease, may be associated with agitation, changes in personality, delusions and restlessness. Asterixis may be transient. Fetor hepaticus is usually present.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


