Alcoholics show genuine thick macrocytes, which are probably related to the toxic effect of alcohol on the bone marrow. Folic acid and vitamin B12 deficiency may contribute.
Bone marrow of chronic hepatocellular failure is hyperplastic and normoblastic. In spite of this, erythrocyte volume is depressed and the marrow therefore does not seem able to compensate completely for the anaemia (relative marrow failure).
Folate and Vitamin B12 Metabolism
The liver stores folate and converts it to its active storage form, tetrahydrofolate. Folate deficiency may accompany chronic liver disease, usually in the alcoholic. This is largely due to dietary deficiency. Serum folate levels are low. Folate therapy is useful. The liver also stores vitamin B12 [4]. Hepatic levels are reduced in liver disease. When hepatocytes become necrotic the vitamin is released into the blood and high serum B12 levels are recorded. This is shown in hepatitis, active cirrhosis and with primary liver cancer. Values in cholestatic jaundice are normal.
Megaloblastic anaemia is rare with chronic liver disease and vitamin B12 therapy is rarely needed.
Erythrocyte Survival and Haemolytic Anaemia
Increased red cell destruction is almost constant in hepatocellular failure and jaundice of all types [5]. This is reflected in erythrocyte polychromasia and reticulocytosis. The mechanism is extremely complex. The major factor is hypersplenism with destruction of red blood cells in the spleen. Also, spur cells have membrane defects, particularly decreased fluidity, and this, with the altered architecture, exacerbates splenic destruction. In some instances, however, the spleen is not the site of erythrocyte destruction. Splenectomy or corticosteroid therapy have little effect [5].
Haemolysis may occur in Wilson’s disease (Chapter 27), and this diagnosis is likely in the young patient presenting with haemolysis and liver dysfunction.
Haemolysis may be acute in patients with alcoholic hepatitis who also have hypercholesterolaemia (Zieve’s syndrome) [6].
Rarely, an autoimmune haemolytic anaemia with a positive Coombs’ test is seen in chronic hepatitis, primary biliary cirrhosis and primary sclerosing cholangitis. Haemolytic anaemia may also follow liver transplantation due to ‘passenger lymphocytes’ in a mismatch donor organ [7] or a delayed transfusion reaction. A syndrome of haemolysis, elevated liver enzymes and a low platelet count (the HELLP syndrome) is a rare complication of the third trimester of pregnancy (Chapter 30) [8]. Haemolysis is a complication of ribavirin therapy due to oxidative damage to the red cell membrane with binding of specific IgG [9].
Aplastic anaemia is a rare complication of acute viral hepatitis, usually type non-A to E hepatitis. It may be fatal but response to intensive immunosuppressive treatment is reported [10]. It may follow liver transplantation [11].
Changes in the Leucocytes and Platelets
Leucopenia and thrombocytopenia are commonly found in patients with cirrhosis, usually with a mild anaemia (‘hypersplenism’).
Leucocytes
The leucopenia is of the order of 1.5–3.0 × 109/L, with the depression mainly affecting polymorphs. Occasionally it may be more severe.
Leucocytosis accompanies cholangitis, fulminant hepatitis, alcoholic hepatitis, hepatic abscess and malignant disease. Atypical lymphocytes are found in the peripheral blood in viral infections such as infectious mononucleosis and viral hepatitis.
Platelets
Abnormalities in platelet count and function are common in patients with all forms of liver disease.
Platelet Count.
In patients with chronic liver disease and portal hypertension, a low platelet count is due in part to increased splenic sequestration and to low thrombopoietin levels. Thus, although platelet counts rise after the insertion of a transjugular intrahepatic portosystemic shunt, they do not return to normal [12]. Plasma concentration of thrombopoietin, the key regulator of platelet function produced mainly by the liver, is reduced in patients with cirrhosis, correlates with platelet count and rises after liver transplantation [13–15].
In chronic liver disease, increased destruction of platelets is minimal and their half-life is normal, calling into question whether there is any biological effect of the IgG and IgM antibodies detected in patients with chronic hepatitis [16,17]. Decreased production of platelets from the bone marrow follows alcohol excess, folic acid deficiency and viral hepatitis.
Platelet Function.
In particular, aggregation is impaired in patients with cirrhosis, particularly Child’s grade C, due to an intrinsic defect and circulating serum factors [18]. There is reduced availability of arachidonic acid for prostaglandin production, and also a reduction in platelet adenosine triphosphate and 5-hydroxytryptamine [19]. Abnormal platelet aggregation due to disseminated intravascular coagulation may be an additional important factor in severe liver failure.
The thrombocytopenia of chronic liver disease (usually 60–90 × 109/L) is extremely frequent and is largely due to hypersplenism. It is very rarely of clinical significance. Unless the patient is actually suffering from the leucopenia or thrombocytopenia the spleen should not be removed; mere demonstration of a low platelet or leucocyte count is not sufficient. The circulating platelets and leucocytes, although in short supply, are functioning well, in contrast to those of leukaemia. Splenectomy is contraindicated. The mortality in patients with liver disease is high and the operation is liable to be followed by splenic and portal vein thrombosis, which preclude later operations on the portal vein and may make hepatic transplantation more difficult.
The Liver and Blood Coagulation [20–22]
Disturbed blood coagulation in patients with hepatobiliary disease is particularly complex [23]. This is due to the many changes in pathways that lead to fibrin production occurring at the same time as changes in the fibrinolytic process (Fig. 4.2, Table 4.1). Changes in platelet number and function are discussed in the previous section. Despite the complexity of the changes, the end result is abnormal coagulation, which needs therapeutic intervention if there is bleeding or if a procedure is planned that risks haemorrhage. However, there is little relationship between abnormal clotting tests and risk of bleeding [24]. Platelet number and function may be more important than the degree of abnormality of the prothrombin time for risk of bleeding with invasive procedures [25].
Table 4.1. Effect of liver disease on haemostasis
| Reduced synthesis of clotting factors |
| hepatic dysfunction per se |
| vitamin K deficiency/ malabsorption |
| Reduced synthesis of inhibitors of coagulation |
| Production of abnormal/ dysfunctional proteins |
| Enhanced fibrolytic activity |
| reduced clearance of activators of fibrinolysis |
| reduced production of inhibitors of fibrinolysis |
| Reduced hepatic clearance of activated clotting factors |
| Disseminated intravascular coagulation |
| multifactorial including endotoxaemia |
| Platelet abnormalities |
| number |
| function |
Fig. 4.2. Normal pathways of (a) coagulation and (b) fibrinolysis. Liver disease effects virtually all components. PAI, plasminogen activator inhibitor; AP, antiplasmin.
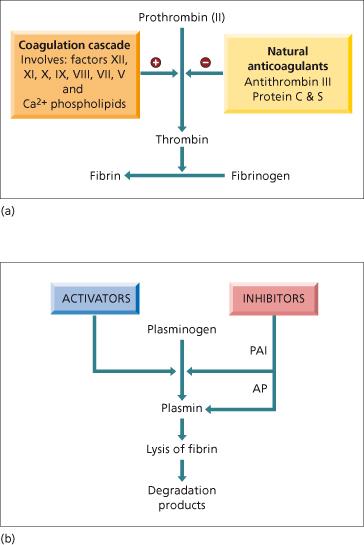
The hepatocyte is the principal site of synthesis of all the coagulation proteins with the exception of von Willebrand factor and factor VIIIC. The proteins include the vitamin K-dependent factors II, VII, IX and X, also labile factor V, factor VIII, contact factors XI and XII, fibrinogen and fibrin-stabilizing factor XIII. The half-life of all these clotting proteins is very short and hence reductions can rapidly follow acute hepatocellular necrosis. Factor VII is particularly affected with a half-life of 100–300 min.
Vitamin K is a fat-soluble vitamin produced by intestinal bacteria. Deficiency occurs most commonly due to cholestasis, intra- and extrahepatic, but may also follow treatment with bile acid chelators (cholestyramine) or oral antibiotics. The vitamin K-dependent proteins are made in the rough endoplasmic reticulum. They all have a number of glutamic acid residues in their aminoterminal region that must be converted, postribosomally, to γ-carboxyglutamic acid by a carboxylase that requires vitamin K [26]. The function of these blood-clotting proteins depends on this conversion since γ-carboxyglutamic acid is responsible for calcium binding, interaction with membrane phospholipids and protease activity. In cholestasis parenteral replacement of vitamin K corrects the prothrombin time (PT) rapidly to normal (24–48 h) and is useful diagnostically. If the coagulopathy is due to hepatic disease the PT may improve but not to normal.
Inhibitors that modulate the coagulation cascade are also synthesized by the liver. These include antithrombin III (ATIII), protein C and S, and heparin co-factor II. Protein C and S are vitamin K dependent. In fulminant hepatic failure [27] and cirrhosis [28] these inhibitors are reduced but their deficiency is paralleled by deficiency of procoagulant factors, so that thrombin generation and thus clot formation is normal or increased (providing there are sufficient platelets) [29]. Congenital deficiencies of coagulation factors such as homozygous protein C deficiency are cured by hepatic transplantation [30].
In liver disease, structurally and functionally inadequate clotting factors and proteins may be produced. Dysfibrinogenaemia is particularly frequent in cirrhosis, chronic hepatitis and acute liver failure. The fibrinogen may contain an excessive number of sialic acid residues. These are thought to lead to abnormal polymerization of fibrin monomers. There may also be a low molecular weight fibrinogen. Abnormalities of fibrinogen account for the prolongation of thrombin time in many patients with liver disease. This should be suspected if the partial thromboplastin time (PTT) is increased but fibrinogen levels are normal and fibrinogen degradation products not increased.
There is evidence for enhanced fibrinolytic activity in patients with liver disease (Fig. 4.2b). Goodpasture first described the accelerated lysis of incubated clotted blood taken from cirrhotic patients in 1914. The synthesis of tissue plasminogen activator by vascular endothelial cells is stimulated in vitro by plasma from patients with decompensated cirrhosis [31]. Hepatocytes synthesize plasmin inhibitors such as α2-antiplasmin, as well as tissue plasminogen activator inhibitor (PAI). In patients with cirrhosis, PAI antigen is reduced even without features of clotting activation (increased fibrin/ fibrinogen degradation products; D-dimer) [18]. The resulting increased tissue plasminogen activator activity relative to PAI activity and α2-antiplasmin is thought to lead to increased fibrinolysis [32],but the relationship is not uniform [33]. Patients with severe liver disease and markers of hyperfibrinolysis are at higher risk of bleeding [34].
Whether there is background disseminated intravascular coagulation (DIC) in patients with cirrhosis, chronic hepatitis and acute hepatitis has been debated for some time [35]. The complex changes found in coagulation proteins, inhibitors and protein fragments usually associated with DIC could have been due to liver disease. Studies of thrombin–antithrombin (TAT) complexes, soluble fibrin, fibrin and fibrinogen degradation products (D-dimer, D-monomer) suggest that low-grade DIC is a component of the coagulopathy in some patients with severe liver disease [36]. The mechanisms stimulating this are thought to include impaired clearance of activated clotting factors, and endotoxaemia [37].
Whatever the background state, cirrhotic patients are, however, at greater risk of overt DIC than patients with normal liver function, particularly in the presence of sepsis and hypotension.
Ascitic fluid contains fibrin monomers, fibrin degradation products and low levels of fibrinogen. This indicates active intraperitoneal coagulation. Fibrinolysis, induced by infusion of plasminogen activators, accounts for the coagulopathy which complicates intravenous infusion of ascitic fluid as in the LeVeen shunt.
Thrombotic complications can be increased in cirrhotic patients [38]. The relationship between antiphospholipid antibodies (lupus anticoagulant, anticardiolipin antibodies) reported in cirrhotic patients [15], the reduction in physiological anticoagulants (ATIII, protein C and S) and the occurrence of thrombotic events remains to be established, but increased thrombin generation has been shown [39], as well as increased risk of deep vein thrombosis [40].
Tests of Coagulation
The PT before and after 10 mg vitamin K given intravenously is the most satisfactory test for a coagulation defect in patients with hepatobiliary disease. It is also a most sensitive indication of hepatocellular necrosis and/or prognosis. The PTT is sometimes performed and is slightly more sensitive than the PT. Prolongation indicates not only deficiency of the prothrombin complex but also factors XI and XII.
Estimation of individual clotting factors is rarely necessary although in patients with fulminant hepatic failure the level of factor V is related to outcome. Thus, in patients with paracetamol-induced (acetaminophen-induced) hepatic failure a factor V concentration of less than 10% on admission predicts a poor outcome [41]. The ratio of factor VIII (increased in liver disease) to factor V on admission is also valuable.
A platelet count less than 50 × 109/L is often associated with abnormal thrombin generation and thus poor clot formation. Measurement of the bleeding time assesses the contribution of platelet number and function to primary haemostasis but does not have a close relationship to bleeding time, contrary to that found in thrombocytopenia due to bone marrow disorders [42].
Fibrinolysis and DIC are diagnosed by marked prolongation of the PT, fibrinogen levels below 1.0 g/L, fibrin degradation products greater than 100 µg/L and thrombocytopenia less than 100 × 109/L.
Thromboelastography (TEG) is a measure of the net outcome of many factors in haemostasis and results are abnormal in cirrhotic patients, particularly when associated with sepsis [43]. Hypocoagulability as measured by TEG may relate to variceal haemorrhage [44]. It is a good test for fibrinolysis.
Management of Coagulation Defect
Vitamin K1 should be given to all patients with a prolonged PT. The usual course is 10 mg vitamin K1 by intravenous injection for 3 days. This is effective in about 6 h and will correct hypoprothrombinaemia related to malabsorption of vitamin K secondary to bile salt deficiency. Defects predominantly due to hepatocellular disease will not be restored by the vitamin K1 treatment. Nevertheless, even in patients with predominantly hepatocellular jaundice there may be a component of bile salt secretory failure and the PT often improves by a few seconds.
A prolongation of the PT of more than 3 s (INR 1.2) after intravenous vitamin K1 has been used as a cut off above which invasive procedures such as percutaneous liver biopsy (but this is not necessary for transjugular biopsy), splenic venography, percutaneous cholangiography or laparotomy should not be performed unless essential, and then under cover of coagulation factors which are effective for a few hours (Table 4.2). However, even patients with PTs and platelet counts regarded as acceptable for invasive procedures (PT <17 s; platelets >80 × 109/L) may have a prolonged bleeding time [42], particularly if liver function is poor. Multiple linear regression analysis shows bleeding time to correlate independently with serum bilirubin concentration and platelet count.
Table 4.2. Routine before invasive techniques (including surgery)
| Measure | Prothrombin time |
| Partial thromboplastin time | |
| Platelet count | |
| Routine | Abstain from alcohol for 1 week |
| Vitamin K1 10 mg intramuscularly | |
| If necessary | Fresh frozen plasma |
| Platelet infusion |
In general, apart from vitamin K1 therapy, it is not necessary to restore blood coagulation to normal in patients with liver disease unless there is active bleeding. Stored blood transfusion will supply prothrombin and factors VII, VIII and X. Fresh blood also supplies factor V and platelets. Fresh frozen plasma is a good source of clotting factors, especially factor V. Recombinant factor VIIa corrects the PT in cirrhotic patients [45,46], but may be insufficient to prevent bleeding—thus its use as a sole agent is not recommended.
Desmopressin (DDAVP), a vasopressin analogue, causes transient shortening of the bleeding time and PTT (but not PT) with increases in factor VIII and von Willebrand factor. However, there is no evidence that it improves haemostasis in vivo.
DIC is treated by control of trigger factors such as infection, shock and dehydration. Fresh blood is most useful but, if unavailable, fresh frozen plasma and packed red blood cells may be used. DIC is never severe enough to merit heparin therapy.
Platelet-rich plasma concentrates are used if thrombocytopenia is a problem and may be given to cover an invasive procedure, but are not needed for such as transjugular liver biopsy or diagnostic ascitic taps.
Hepatic Transplantation
The bleeding problems associated with liver transplantation are mainly due to hyperfibrinolysis during the an hepatic phase superimposed on the coagulopathy due to the underlying end-stage liver disease. In the early era of liver transplantation operative blood loss often led to replacement with about 20 units of red blood cells and 15 units of platelets. Surgical skill and experience are important factors in reducing blood loss and liver transplant is reported without use of any blood transfusion [47]. Prognosis is related to the volume of blood and blood products given [48].
During surgery, coagulation and fibrinolysis are activated. Activation of the fibrinolytic system occurs particularly during the anhepatic and postreperfusion stages. Plasma concentrations of tissue-type plasminogen activator increase and there are high concentrations of fibrin degradation products.
Aprotinin, a low molecular weight serine–protease inhibitor with potent antifibrinolytic activity, reduces blood transfusion requirements during liver transplantation by 50–60% [49]. There is no increased complication rate from thrombus formation. Another antifibrinolytic agent, tranexamic acid, is now used to reduce blood loss during liver transplantation [50], as aprotinin has caused increased thrombosis in patients with cardiac surgery, a finding however not seen in liver transplantation [51].
Haemolytic Jaundice
Haemoglobin is released in excessive amounts, increasing from the normal of 6.25 g to as much as 45 g daily. Consequently, there is an increase in the serum bilirubin, 85% of which is unconjugated. The rise in conjugated bilirubin is probably due to reflux from hepatocytes.
Even if bilirubin production reaches its maximum of 1500 mg daily (six times normal), serum bilirubin rises only to about 2–3 mg/100 mL (35–50 µmol/L). This is because of the great capacity of the liver to handle bilirubin. If patients with haemolytic jaundice show serum bilirubin values greater than 70–85 µmol/L there is probably the additional factor of Gilbert’s syndrome, hepatocellular dysfunction or kidney failure. Anaemia itself will, of course, depress liver function.
Unconjugated bilirubin is not water soluble and does not pass into the urine. A little bilirubin may be detected in the urine by sensitive tests if the conjugated level in the blood rises to values that are unusually high for haemolysis.
Bile pigment excretion is greatly increased and large quantities of stercobilinogen are found in the stools. Each milligram of stercobilinogen corresponds to the breakdown of 24 mg haemoglobin. This estimate can only be approximate, for a significant proportion of the faecal haem pigment is derived from sources other than haemoglobin of mature erythrocytes.
Pathological Changes
The breakdown of haemoglobin yields iron. Tissue siderosis is a feature of most types of haemolytic anaemia.
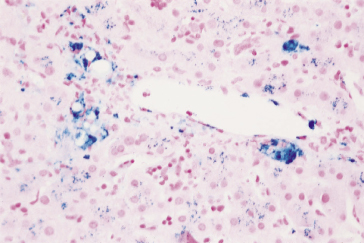
The liver is normal sized and is reddish-brown due to increased amounts of iron. Histology shows iron in the Kupffer cells, large macrophages of the portal tracts and, to a lesser extent, in hepatocytes (Fig. 4.3). In advanced stages, the accumulation in the hepatocytes increases and the appearance becomes indistinguishable from primary haemochromatosis. In the severely anaemic, there is centrizonal sinusoidal distension with fatty change. The Kupffer cells are generally swollen and hyperplastic foci of erythropoiesis are uncommon. The gallbladder and bile passages contain dark viscid bile. Calcium bilirubinate pigment calculi are found in one-half to two-thirds of patients.
Fig. 4.3. Hepatic siderosis due to haematological disease. Increased amounts of iron (stained blue) are seen in the larger Kupffer cells and portal macrophages and, to a lesser extent, as granular staining in hepatocytes. (Perls’ stain.)
The spleen is enlarged, fleshy and packed with erythrocytes. The red bone marrow is hyperplastic.
Clinical Features
The picture varies with the cause, but certain symptoms and signs are common to all forms of haemolysis.
Anaemia depends on the rate of destruction compared with regeneration of red blood cells. The haemoglobin falls rapidly with crises where the patient becomes ill with aching pains in the abdomen and limbs, fever, headache and sometimes even a fall in blood pressure and collapse.
Jaundice is usually mild and lemon yellow. It increases rapidly with haemolytic crises or if there is a coincidental difficulty in biliary excretion such as viral hepatitis or choledocholithiasis or if the kidney fails.
Bilirubin pigment gallstones may be associated with the features of chronic cholecystitis. Stones in the common bile duct may cause obstructive jaundice, and the coexistence of two types of jaundice provides a confusing clinical picture. Gallstones in children always suggest a haemolytic aetiology.
Splenomegaly is present in the chronic forms.
Ulcers or pigmentation from healed ulcers, usually over the internal or external malleoli, occur in some types.
Haematological Changes
Anaemia is variable and the peripheral blood shows a reticulocytosis. Leucocytes are usually increased.
The bone marrow is hyperplastic and the proportion of erythroid to leucopoietic cells rises.
The survival of labelled erythrocytes is reduced and increased uptake can be shown in the spleen.
In some hereditary anaemias iron overload may occur without transfusion. This is particularly when there is a high degree of ineffective erythropoiesis, for example in congenital dyserythropoietic anaemias, congenital sideroblastic anaemia and thalassaemia intermedia. It may also occur in pyruvate kinase deficiency [52]. Rarely, concurrent presence of mutations in Hfe gene may accentuate iron deposition [53] but generally do not appear to be a prerequisite for the iron deposition (Chapter 26).
Faeces and Urine
The faeces are dark and stercobilinogen is increased. Urobilinogen is increased in the urine. Bilirubin is detected in the urine only rarely, when jaundice is deep. When red cell destruction is rapid, free haemoglobin may be found in the urine and microscopy reveals pigmented casts.
Serum Biochemistry
Serum unconjugated bilirubin levels are raised but conjugated bilirubin is only slightly increased. The serum alkaline phosphatase, albumin and globulin concentrations are normal. Serum haptoglobins are diminished. The serum cholesterol level is low.
If haemolysis is particularly acute, methaemalbumin can be detected in the serum. Serum ferritin is increased. Free haemoglobin may be detected.
Differential Diagnosis
The diagnosis of haemolytic from other forms of jaundice is usually easy. The absence of pain, pruritus, the dark colour of the stools and normal alkaline phosphatase are points of difference from cholestatic jaundice. The absence of stigmas of hepatocellular disease, the normal serum alanine transaminase and protein values distinguish it from viral hepatitis and cirrhosis.
Distinction from the congenital unconjugated hyperbilirubinaemias may be difficult, particularly as many patients with Gilbert’s disease show a decreased erythrocyte survival.
The Liver in Haemolytic Anaemias
Hereditary Spherocytosis [54]
The main signs are jaundice, anaemia, splenomegaly and gallstones, but the spectrum of disease is wide, from no clinical expression to death in utero. Inheritance is dominant or recessive. In 70% of cases the molecular defect is a mutation in ankyrin, one of the components of the red cell skeleton.
Jaundice is rarely noticed before school age or adolescence. The mean serum bilirubin level is 35 µmol/L (2 mg/dL) (range 10–100 µmol/L). Deep jaundice is rare. This may develop in the neonatal period and be associated with incipient kernicterus.
Gallstones are related to age and are rare at less than 10 years of age. They are symptomatic in about half of the patients. The stones are usually removed at the time of splenectomy.
Hereditary elliptocytosis, another genetic defect due to a mutation in a protein within the red cell membrane skeleton, is usually a harmless trait, the haemolysis being compensated. It may occasionally develop into active decompensated haemolytic anaemia.
Various Enzyme Defects
Many of the hereditary non-spherocytic anaemias are now known to be due to various defects in the metabolism of the red cells. They include deficiency of pyruvate kinase or triose phosphate isomerase, or deficiency in the pentose phosphate pathway such as glucose-6-phosphate dehydrogenase (G6PD). These conditions may be of particular importance in the aetiology of neonatal jaundice. The gene responsible for G6PD deficiency has now been cloned and a wide range of mutations recognized. These are beginning to explain the wide spectrum of clinical pictures seen in this condition ranging from haemolysis during the neonatal period, after infection or after the ingestion of certain drugs, to chronic anaemia irrespective of any of these factors. Variants of the gene are now recognized where there is no significant reduction in enzyme activity in red cells [55].
Viral hepatitis can precipitate destruction of G6PD-deficient cells and so cause acute haemolytic anaemia and very high serum bilirubin concentrations.
Sickle Cell Disease [56,57]
The abnormal haemoglobin crystallizes in the erythrocytes when the oxygen tension is reduced. There are crises of blood destruction with acute attacks of pain. The liver may be affected acutely by sickling crises. There is right upper quadrant pain, fever and increased jaundice, associated with systemic and haematological features of sickling. This should help to differentiate the clinical picture from a common bile duct stone. Fulminant liver failure is rare [58]. A distinct clinical picture of intrahepatic cholestasis is also recognized but is unusual [59]. Histologically there is intracanalicular cholestasis, sinusoidal dilatation, packing of the sinuses by sickled erythrocytes, Kupffer cell hyperplasia and erythrophagocytosis.
There may be chronic elevation of transaminases and/or alkaline phosphatase with hepatic scarring. Several factors have been implicated including microvascular stasis, with recurrent ischaemic episodes, and transfusion-related disease (haemosiderosis and viral hepatitis).
Jaundice accompanying sickle cell disease is always particularly deep, the high serum bilirubin levels being related to the combination of haemolysis and impaired hepatocellular function. Depth of jaundice per se should not be regarded as an indication of severity. Concomitant viral hepatitis or obstructed bile ducts lead to exceptionally high serum bilirubin values.
Gallstones are found in 25% of children and 50–70% of adults with homozygous sickle cell disease. They are usually in the gallbladder; duct calculi are rare. In two-thirds of adults the stones are asymptomatic. The high frequency of gallbladder stones may be due in part to changes in gallbladder volume and motility. Elective cholecystectomy may be hazardous and precipitate a sickle crisis [56].
Hepatic Histology
Active and healed areas of necrosis may have followed anoxia due to vascular obstruction by impacted sickle cells or by Kupffer cells swollen with phagocytosed erythrocytes following intrahepatic sickling. The widened sinusoids show a foam-like fibrin reticulum within their lumen. This intrasinusoidal fibrin may later result in fibre deposition in the space of Disse and narrowed sinusoids. Bile plugs are prominent. Fatty change is related to anaemia. Multiple transfusions lead to hepatic siderosis which is not accurately reflected by the serum ferritin [60].
The classic findings are of intrasinusoidal sickling, Kupffer cell erythrophagocytosis and ischaemic necrosis. It is difficult to explain the severe liver dysfunction on these histological findings, which have been reported largely on autopsy specimens. Superimposed complications such as septicaemia or viral hepatitis complicate the histological findings [61].
Electron Microscopy
The changes are those of hypoxia. There are sinusoidal aggregates of sickled erythrocytes, fibrin and platelets, with increased collagen and occasional basement membrane-like material in the space of Disse.
Clinical Features
Asymptomatic patients commonly have raised serum transaminases and hepatomegaly. Hepatitis B and C and iron overload may have complicated transfusions.
In about 10%, the crisis selectively affects the liver. It lasts 2–3 weeks. It is marked by abdominal pain, fever, jaundice, an enlarged tender liver and a rise in serum transaminases. In some patients the crisis is precipitated by Salmonella infection or by folic acid deficiency.
Acute liver failure, usually with cholestasis, is rare. Jaundice is very deep with a markedly increased PT and encephalopathy but with only modestly increased serum transaminases. Liver biopsy shows the changes of sickle cell disease with marked zone 2 necrosis and cholestasis. The diagnosis of hepatic sickle crisis from viral hepatitis is difficult. In general, in viral hepatitis pain is less, jaundice deeper and transaminase elevations more prolonged. Liver biopsy and hepatitis viral markers usually help to make the distinction. Exchange transfusion has been successful [58]. Liver transplantation has been attempted successfully in carefully selected patients [62].
Prolonged intrahepatic cholestasis associated with sickle cell anaemia has also responded to exchange transfusion [59].
Acute cholecystitis and choledocholithiasis may simulate hepatic crisis or viral hepatitis. Magnetic resonance, endoscopic or percutaneous cholangiography are important investigations in excluding biliary obstruction. Complications after cholecystectomy are common, and this is indicated only if there is great difficulty in making a distinction from abdominal crisis or where symptoms are clearly related to gallbladder disease. Preoperative exchange transfusion may lessen later complications.
General features include leg ulcers, which are frequent. The upper jaw is protuberant and hypertrophied. The fingers are clubbed. Bone deformities seen radiologically include rarefaction and narrowing of the cortex of the long bones and a ‘hair-on-end’ appearance in the skull.
Thalassaemia
Crises of red cell destruction and fever and the reactionary changes in bone are similar to those seen in sickle cell disease. The liver shows siderosis and sometimes fibrosis. The haemosiderosis may progress to an actual haemochromatosis and require chelation therapy using desferrioxamine or other chelator therapy (Chapter 26). The stainable iron in the liver cells may be greater in those who have undergone splenectomy (usually performed to reduce blood transfusion requirements) as a storage organ for iron.
Transfusion-acquired hepatitis B and C may lead to chronic liver disease.
Episodes of intrahepatic cholestasis of uncertain nature can also develop. Gallstones may be a complication.
Previously, the commonest cause of death in thalassaemia major was heart failure but the clinical course of the disease is changing with improved therapy including, in particular, iron chelation.
Treatment
This may include folic acid, blood transfusion, iron chelation therapy, antiviral treatment and occasionally splenectomy with pneumococcal vaccination. Bone marrow transplantation may be considered but the survival is worse in those with liver disease [63].
Paroxysmal Nocturnal Haemoglobinuria [64]
In this rare acquired disease, there is intravascular, complement-mediated haemolysis. The defect is due to mutation of the PIG-A
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


