It is important to differentiate between those diseases that are self limiting, respond to therapy or require early surgery such as biliary atresia.
Clinical Features and Diagnosis (Table 29.4) [15]
Conjugated hyperbilirubinaemia may present at any time after birth. If it is detected in the first 24 h of life, sepsis must be excluded. Most causes of the neonatal liver disease have a similar presentation. The clinical history should include:
- details about the mother’s pregnancy (drugs, alcohol, smoking, intercurrent illnesses, pruritis of pregnancy, hepatitis status and risk factors, e.g. drug abuse)
Table 29.4. Conjugated hyperbilirubinaemia in neonates
| Infection |
| Viruses (cytomegalovirus, rubella, Coxsackie, herpes simplex) (Chapters 21) |
| Syphilis |
| Toxoplasmosis |
| Bacteria (Escherichia coli) |
| Metabolic |
| Galactosaemia |
| α1-Antitrypsin deficiency |
| Tyrosinaemia type 1 |
| Cystic fibrosis |
| Hereditary fructose intolerance |
| Total parenteral nutrition |
| Niemann–Pick C disease |
| Progressive familial intrahepatic cholestasis 1,2,3 |
| Alagille’s syndrome |
| Endocrine |
| Hypopituitarism |
| Hypothyroidism |
| Biliary atresia |
| Choledocal cyst |
| Miscellaneous |
| Inspissated bile syndrome (erythroblastosis with cholestasis) |
| Vascular causes |
| ‘shock’ |
| congenital heart disease |
- Birth weight and gestational age
- Vitamin K administration
- Family history and consanguinity.
History of the present illness should include:
- Date of jaundice
- Colour of stools and urine
- Drug history, particularly parenteral nutrition
- Bleeding
- Petechiae or bruising
- Feeding history and weight gain
- Diarrhoea and vomiting.
The clinical features include:
- Jaundice, dark urine, pale yellow stools, failure to thrive or poor feeding
- Infants may be small for gestational age, especially those with Alagille’s syndrome, metabolic liver disease and intrauterine infection
- Dysmorphic features in trisomy 18, trisomy 21, Alagille’s syndrome, Zellweger’s syndrome, and with certain congenital infections
- Hypoglycaemia in metabolic liver disease, hypopituitarism or severe liver disease
- Hypogonadism (in males) and optic dysplasia in hypopituitarism
- Hepatomegaly with or without splenomegaly
- Ascites is rare except in metabolic liver disease
- Bleeding from vitamin K deficiency (particularly if breast fed) or thrombocytopenia.
Investigations
Findings from biochemical liver function tests in conjugated hyperbilirubinaemia include:
- Total serum conjugated hyperbilirubin exceeds 20 µmol/L (1.2 mg/dL).
- Serum aminotransferases are usually elevated two to four times normal. Higher elevations suggest an infectious process.
- Serum alkaline phosphatase may be normal or only mildly elevated. High levels may indicate biliary obstruction or rickets.
- Serum γ-GT may be elevated. Normal or low γ-GT suggests certain bile canalicular transporter defects (Byler disease or progressive familial intrahepatic cholestasis).
- Serum albumin is usually normal unless there is severe prenatal disease.
- Serum cholesterol is usually elevated in children with severe cholestasis, for example in Alagille’s syndrome or biliary atresia.
- Prothrombin and partial thromboplastin times are usually normal unless there is associated vitamin K deficiency (haemorrhagic disease of the newborn) or severe liver disease.
- Serum α1-antitrypsin level and phenotype for the diagnosis of α1-antitrypsin deficiency.
Serology for TORCH Screen.
Antibodies to herpes simplex, rubella, Toxoplasma, cytomegalovirus, adenovirus and Coxsackie viruses are estimated in both baby and mother and serology for syphilis. The serum is usually tested for hepatitis B surface antigen (HBsAg), IgM antihepatitis B core antigen (anti-HBcAg), IgM antihepatitis A virus (anti-HAV), antihepatitis C virus (anti-HCV) and HCV RNA, but these are rare causes of neonatal liver disease.
Urine Tests.
Cultures are taken for Gram-negative organisms and for cytomegalovirus infection. Aminoaciduria is noted. Reducing substances are sought if galactosaemia is suspected.
Metabolic Tests.
These include urinary reducing substances; the red cell enzyme galactose-1-phosphate uridyl transferase, urine and plasma amino acids and organic acids.
Endocrine Tests.
These include thyroid function tests and a 09.00 h cortisol level.
Bile Salts.
Urinary bile salts for assessment by fast atom bombardment mass spectrometry and tandem mass spectrometry may be required to identify primary bile salt deficiencies.
Other Specific Tests.
These include measurement of carnitine and acyl carnitine in fatty acid oxidation disorders.
Abdominal Ultrasound Scan.
This may be carried out (after 4-h fast) to detect gallbladder size. The gallbladder is usually present unless there is severe intrahepatic cholestasis or biliary atresia. Ultrasound can also diagnose choledochal cyst.
Radioisotope Scan.
This may be used to demonstrate hepatic uptake and biliary excretion (which may be delayed more than 4–6 h in neonatal hepatitis syndrome if there is severe cholestasis, and more than 24 h in biliary atresia).
Liver Biopsy.
Needle biopsy of the liver is easy and well tolerated in neonates, infants and children. It is required to determine the severity of hepatocellular injury and extent of fibrosis, evidence for infiltrative or storage disease and to differentiate intrahepatic cholestasis from extrahepatic obstruction. Portal zone duct proliferation and a biliary type of fibrosis are usually present in extrahepatic biliary atresia (Fig. 29.1b). A relative paucity of portal zone bile ducts supports the diagnosis of intrahepatic cholestasis. The PAS-positive bodies of α1-antitrypsin deficiency may be seen after 2 months. Electron microscopy is essential if metabolic disease is suspected.
Percutaneous and Endoscopic Cholangiography.
The percutaneous technique is of value when liver biopsy findings are equivocal and the TeBIDA suggests biliary atresia. Endoscopic cholangiography is employed using suitably sized instruments [16].
Family Studies.
At the onset it is valuable to test the blood of the mother, father and other siblings by appropriate methods and to store the sera for later use, or to collect DNA to examine for genetic diseases.
Neonatal Hepatitis Syndrome
This may be due to: (1) intrauterine infections, (2) endocrine causes such as hypothyroidism, or (3) inherited diseases including chromosomal abnormalities.
Intrauterine Infection
Immunity is reduced in the neonate and virus infections are frequent and very liable to persist. Chronic hepatitis and cirrhosis may ensue. Similarly, older children with immunological deficits such as agammaglobulinaemia or who are receiving treatment with immunosuppressive drugs are at risk.
Toxoplasmosis, Rubella, Cytomegalovirus, Herpes Simplex (TORCH) Infections
Congenital infections grouped under the acronym TORCH have similar clinical features: hepatosplenomegaly, jaundice, pneumonitis, petechial or purpuric rash, prematurity or poor intrauterine growth. Presentation with acute liver failure may occur with herpes simplex infection.
Cytomegalovirus
This is the commonest cause of intrauterine infection (Chapter 21). It is usually acquired placentally from an asymptomatic mother. It can also be transmitted in breast milk and from blood products. Many congenital infections are asymptomatic.
The disease may be fulminant with intense jaundice, purpura, hepatosplenomegaly, chorioretinitis, cataracts and pulmonary defects. Survivors may run a long course with persistent jaundice, hepatomegaly and disappearing bile ducts. The prognosis is good although 30% will develop cirrhosis requiring treatment by liver transplantation.
Intranuclear viral inclusions are seen in bile duct epithelium and rarely in hepatocytes. Diagnosis is made on urine or tissue in situ using PCR [17].
Herpes Simplex
The liver may be involved in the course of a fulminating viraemia, contracted at birth from maternal genital herpes. Jaundice is due to viral involvement of the liver. Histologically, necrosis is seen with little or no inflammatory reaction. Giant cells are absent, but inclusion bodies may be found.
Treatment with i.v. ganciclovir is essential. Liver transplantation is usually contraindicated if there is multiorgan failure and the mortality is 70% [18].
Congenital Rubella Syndrome
This disease, if contracted in the first trimester of pregnancy, may cause fetal malformations. It may also persist through the neonatal period and into later life. The liver with the brain, lung, heart and other organs are involved in the generalized virus infection. Jaundice develops within the first 1 or 2 days with hepatosplenomegaly, cholestasis and slightly elevated serum transaminase levels.
Hepatic histology shows a typical giant cell hepatitis, with bile in swollen Kupffer cells and ductules with a focal hepatocellular necrosis and portal fibrosis. Erythroid haemopoietic tissue is relatively increased. The virus can be identified from the liver at necropsy or biopsy. Usually the hepatitis resolves completely.
Intrauterine parvovirus B19 can cause severe giant cell hepatic disease in the neonate, also fulminant liver failure and aplastic anaemia [19].
Congenital Toxoplasmosis
Congenital toxoplasmosis is rare and results from maternal infection in the third trimester. Neonatal hepatitis is associated with central nervous system involvement with chorioretinitis (with large pigmented scars), hydrocephaly or microcephaly. The liver shows infiltration of portal zones with mononuclear cells. Extramedullary haemopoiesis with increased stainable iron is conspicuous. Histiocytes containing Toxoplasma may be present. It is diagnosed by finding Toxoplasma IgM antibodies and treated with spiramycin.
Congenital Syphilis
Congenital syphilis is increasing again in frequency [20]. It causes a multisystem illness, with intrauterine growth retardation, failure to thrive, severe anaemia, thrombocytopenia, nephrotic syndrome, periostitis, nasal discharge (‘snuffles’), skin rash, diffuse lymphadenopathy and hepatomegaly. Jaundice may be severe. Central nervous system involvement occurs in up to 30% of infants. Diagnosis involves serological testing, including the Venereal Disease Research Laboratory (VDRL) test and confirmatory testing for specific antitreponemal antibodies. Treatment is with penicillin.
Varicella
Varicella may occur in newborn infants if maternal infection occurs within 14 days of delivery. The disease is characterized by jaundice, extensive skin and multisystem involvement, especially pneumonia. In severe or fatal cases hepatic parenchymal involvement can be demonstrated [21]. Treatment is with aciclovir.
Human Immunodeficiency Virus (HIV) Infection
Congenital HIV infection may present clinically as hepatitis with jaundice although later than in the neonatal period, typically at approximately 6 months of age [22]. Older children have a similar picture to adults with the same spectrum of infections, primary lymphoma and Kaposi’s sarcoma. Hepatic histology shows more giant cell transformation and fewer granulomas [23]. Diffuse, lymphoplasmocytic infiltration is associated with lymphoid interstitial pneumonia.
Hepatitis A, B and C
Infections with these viruses in the neonate do not cause jaundice or neonatal hepatitis unless there is acute liver failure or severe hepatitis (see below).
Bacterial Infection Outside the Liver
In the neonate, an immature reticuloendothelial system with decreased complement and opsonins impairs the ability of the liver and spleen to phagocytose bacteria. Conjugated hyperbilirubinaemia may occur with sepsis or localized extrahepatic infection, such as a urinary infection [24,25]. Serum aminotransferases may be slightly elevated, but hepatosplenomegaly is uncommon. Jaundice may also occur with streptococcal and staphylococcal infections and Gram-negative bacterial septicaemia.
Endocrine Disorders
Hypothyroidism
Hypothyroidism is usually associated with an unconjugated hyperbilirubinaemia or the neonatal hepatitis syndrome and should be excluded in every patient. It is more common in girls than boys. Mild anaemia is common and the infant is sluggish. The diagnosis is confirmed by finding low serum thyroxine and tri-iodothyronine levels with high thyroid-stimulating hormone, and by observing the effects of therapy. The mechanism of the jaundice is unknown.
Hypopituitarism
Pituitary–adrenal dysfunction is associated with neonatal hepatitis syndrome in 30–70% of patients [26]. Clinical features include: conjugated hyperbilirubinaemia; hypoglycaemia in the perinatal period, septo-optic dysplasia [27] and hypoplasia of the optic nerves which is associated with hypopituitarism. There may also be midline facial abnormalities, nystagmus and microgenitalia in boys.
The diagnosis is made by identifying a low thyroid-stimulating hormone and a free thyroxine level with a low 9.00 h cortisol value. It resolves with hormone replacement, thyroxine and hydrocortisone.
Chromosomal Disorders
Trisomy 18
Trisomy 18 is associated with growth retardation, skeletal abnormalities and complex congenital heart disease. A giant-cell hepatitis has been reported [28,29].
Trisomy 21
The association between trisomy 21 and neonatal cholestasis or extrahepatic biliary atresia is reported. The neonatal hepatitis resolves spontaneously.
Idiopathic Neonatal Hepatitis
This is diagnosed after exclusion of known causes. The number of cases being diagnosed has diminished with the developments in molecular and genetic technology.
Neonatal Hepatitis in Preterm Infants
Idiopathic neonatal hepatitis is a common referral in preterm babies, as many more premature infants survive. Most will have been given parenteral nutrition and are at risk of cholestasis. It is important to differentiate this condition from other known causes of neonatal hepatitis syndrome, parenteral nutrition-associated liver disease and biliary atresia which may have an atypical presentation in this age group. Examination of stools for pigment and a fasting ultrasound examination for gallbladder size are useful investigations to exclude biliary atresia. Liver biopsy is only indicated if there is persistent elevation of conjugated bilirubin and/or abnormal liver biochemistry. The prognosis is good.
Prolonged Parenteral Nutrition
The aetiology of parenteral nutrition-related liver disease is complex and is associated with prematurity, low birth weight and recurrent sepsis [30]. It is related to difficulties in enteral feeding, loss of the enterohepatic circulation of bile acids and consequent reduced bile formation, biliary stasis and sludging.
Infants have a gradual increase in serum conjugated bilirubin associated with elevation of transaminases, alkaline phosphatase and γ-GT. Liver biopsy shows non-specific changes with features of extrahepatic biliary obstruction and bile in canaliculi (Fig. 29.1c). Biliary sludge and gallstones may develop. The disease is reversible if parenteral nutrition can be discontinued, but in infants with intestinal failure is the main indication for combined liver and bowel transplantation [30].
Inherited Disease in the Neonate
α1-Antitrypsin Deficiency
α1-antitrypsin is synthesized in the rough endoplasmic reticulum of the liver. It comprises 80–90% of the serum α1-globulin and is an inhibitor of trypsin and other proteases. Deficiency results in the unopposed action of these enzymes, in particular neutrophil elastase. The lungs are the major target, with damage to alveoli and resulting emphysema [31].
The gene for α1-antitrypsin is on chromosome 14. There are about 75 different alleles at this locus, which can be distinguished by isoelectric focusing or agarose gel electrophoresis at acid pH, or by PCR analysis. M is the common, normal allele. Z and S are the most frequent abnormal alleles, which put the individual at risk of disease. One gene is derived from each parent. The combination results in normal, intermediate, low or zero serum α1–antitrypsin levels. Protease inhibitor (Pi) MM gives a serum α1-antitrypsin value of 20–53 µmol/L, the normal state. PiZZ results in a low concentration of 2.5–7 µmol/L and PiNull-Null gives zero levels. Both give a high risk of emphysema. PiSS and PiMZ give levels 50–60% of normal with no increased risk of lung disease. PiSZ gives α1-antitrypsin levels of 8–19 µmol/L with a mildly increased risk [32,33].
Mutation of the gene can give deficiency of circulating α1-antitrypsin by a number of mechanisms. Liver disease, however, only occurs with mutations where α1-antitrypsin accumulates in hepatocytes. The classical type is PiZZ but the Mmalton and Mduarte variants may do the same.
Pathogenesis of Liver Disease [33,34].
Only the PiZZ phenotype has been clearly associated with liver disease. This is not due to the low circulating levels of α1-antitrypsin arriving at the liver, since other phenotypes with a low circulating levels do not develop hepatic damage. Intrahepatic accumulation of α1-antitrypsin seems to be responsible. Studies of the molecular structure have shown that with the ZZ mutation there is polymerization of protein units. Normally, the reactive loop (Fig. 29.2) swings in between the β-helices of the so-called A-sheet of the protein, where it interacts with elastase and other enzymes. In the ZZ mutant protein, the reactive loop cannot do this. It remains on the outside and is then available to insert into the A-sheet of an adjacent ZZ unit [35]. The polymers formed prevent export of most of the protein.
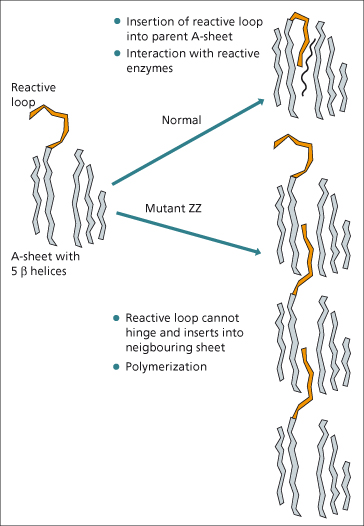
Accumulation of ZZ protein is thought to be responsible for liver damage but the mechanism is still unclear. Polymerization of ZZ protein occurs spontaneously or following minor perturbations such as a rise in temperature. However, the mutation of the α1-antitrypsin protein is not the only reason for its retention. Cells from individuals with α1-antitrypsin liver disease also have a reduction in the degradatory pathways in the endoplasmic reticulum [33]. The variation in clinical disease therefore appears to depend not only on the abnormal protein produced in PiZZ but also other cellular mechanisms as yet poorly understood.
Only a small proportion of individuals with α1-antitrypsin deficiency ever develop liver disease, but it is the main cause of emphysema in early adulthood.
Clinical Picture.
α1-antitrypsin deficiency is the commonest inborn error of metabolism to present with persistent neonatal jaundice, with an incidence of 1 : 7000 live births worldwide. Infants may present with intrauterine growth retardation, cholestasis, failure to thrive, hepatomegaly, or a vitamin K responsive coagulopathy. The latter is more likely in those infants who are not given prophylactic vitamin K at birth and who are being breast fed.
Liver biochemistry demonstrates a mixed hepatic/ obstructive picture with elevated transaminases, alkaline phosphatase and γ-GT. Radiological investigations may indicate severe intrahepatic cholestasis with a contracted gallbladder on a fasting ultrasound, and delayed or absent excretion of radioisotope on hepatobiliary scanning. In homozygotes, the diagnosis is easily confirmed by detection of a low level of a1-antitrypsin (<0.9 g/L).
Liver histology demonstrates giant cell hepatitis with characteristic periodic acid–Schiff (PAS), diastase resistant, positive granules of α1-antitrypsin in hepatocytes, which may be detected by 6–8 weeks of age.
Management consists of nutritional support, fat-soluble vitamin supplementation and treatment of pruritus and cholestasis. Patients and parents should not smoke, and PiZZ individuals should be protected from passive smoking.
Parents are obligate heterozygotes. Thus there is a 25% chance of each subsequent fetus being affected. Antenatal diagnosis by chorionic villus sampling is now available using synthetic oligonucleotide probes specific for the M and Z gene or by restriction fragment length polymorphism [36].
Prognosis.
The prognosis is variable. The long-term outlook for many infants with α1-antitrypsin deficiency is good and in approximately 50% the liver disease resolves; 25% develop chronic liver disease [37], while the remainder require liver transplantation in the first year of life. Poor prognostic factors are prolonged jaundice, a higher AST at presentation and histology with severe bile duct reduplication, severe fibrosis with bridging septa and cirrhosis [38].
The few children with α1-antitrypsin deficiency who present later in infancy or in childhood with hepatomegaly, but without any neonatal jaundice, usually are cirrhotic and have a poor prognosis.
Associations in Later Life.
The incidence of liver disease in PiZZ individuals at age 50 years is about 15%, more frequent in males. Usually the changes in liver function are subtle, but the patient may present with complications of portal hypertension or ascites. Hepatocellular carcinoma may complicate cirrhosis.
Cystic Fibrosis
Abnormalities of liver function tests or hepatic pathology are found in one-third of infants with cystic fibrosis. The spectrum of hepatic disorder is highly variable, but the clinical presentation is with jaundice, hepatomegaly, and failure to thrive and meconium ileus. Some infants have a giant-cell hepatitis. Extrahepatic bile duct obstruction may be due to inspissated bile actually plugging the common bile duct [39], which can be removed by choledochotomy. Occurrence of neonatal hepatitis syndrome in cystic fibrosis does not necessarily predict early development of cirrhosis.
Bile Duct Paucity Syndromes
Alagille’s Syndrome (Arteriohepatic Dysplasia) [40].
This is an autosomal dominant condition with an incidence of 1 : 100 000 live births worldwide. It is a multisystem disorder which is associated with cardiac, facial, renal, ocular and skeletal abnormalities. The condition is related to a deletion on the short arm of chromosome 20p, which has been identified in 60% of patients. It is related to mutations in the Jagged 1 gene, which encodes a ligand of Notch 1, one of four members of a family of transmembrane receptor proteins [41]. The mutation is predominately sporadic [42].
Infants present with persistent cholestasis, severe pruritus, hepatomegaly and failure to thrive. The characteristic facial features are difficult to identify in infancy, but become prominent later in childhood. They include a triangular face with high forehead and frontal bossing, deep widely spaced eyes, saddle-shaped nasal bridge and pointed chin (Fig. 29.3). Cardiac abnormalities include peripheral pulmonary stenosis, pulmonary and aortic valve stenosis and Fallot’s tetralogy [40]. Hepatosplenomegaly is unusual unless there is progressive fibrosis, which is rare.
Fig. 29.3. In Alagille’s syndrome, the characteristic facial features are difficult to identify in infancy, but become prominent later in childhood or in adult life (as in the father). They include a triangular face with high forehead and frontal bossing, deep widely spaced eyes, saddle-shaped nasal bridge and pointed chin.
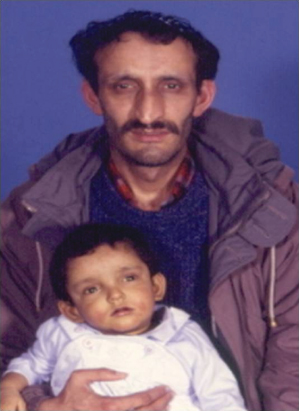
Skeletal abnormalities include abnormal thoracic vertebrae, ‘butterfly’ vertebrae and curving of the proximal digits of the third and fourth finger. Ocular abnormalities [43,44] include optic disease and papilloedema secondary to intra cranial hypertension, while posterior embryotoxin, which is detected on the inner aspects of the cornea near the junction of the iris, is demonstrated in 90% of patients by slit-lamp examination. Renal disease varies in severity from mild renal tubular acidosis to severe glomerular nephritis. Severe failure to thrive, which is complicated by gastrointestinal reflux and severe steatorrhoea secondary to fat malabsorption or pancreatic insufficiency is difficult to treat.
Liver biochemistry indicates severe cholestasis with: conjugated bilirubin above 100 μmol/L (6 mg/dL); raised alkaline phosphatase above 600 IU/L; γ-GT above 200 IU/L; raised transaminases; and plasma cholesterol above 6 mmol/L with normal triglycerides 0.4–2 mmol/L.
Tests of hepatic function such as albumin and coagulation are usually normal. Liver histology may be non-specific. The reduction in interlobular bile ducts is often difficult to identify in the neonatal period, particularly if cholestasis and giant cell hepatitis are also present. The histological appearance differs from extrahepatic biliary atresia because of the absence of portal fibrosis and biliary ductular proliferation.
Management.
Intensive nutritional support is essential and pancreatic supplements may be required. Pruritus may be intractable and an indication for liver transplantation, although recent experience with the Molecular Reabsorbent Recirculating System (MARS) have produced relief for 6–12 months [45].
Prognosis is varied and depends on the extent of liver, cardiac or renal disease. Approximately 50% of children may regain normal liver function by adolescence while others require liver transplantation in childhood [40]. The 20 years predicted life expectancy is 75% for all patients, 80% for those not requiring liver transplantation and 60% for those who require liver transplantation [46]. The indications for liver transplantation are the development of cirrhosis and portal hypertension, intractable pruritus or severe decompensated growth failure. Pretransplant cardiac surgery or balloon dilatation may be indicated for severe pulmonary stenosis.
Genetic Cholestatic Syndromes
These diseases are related to defects in one of the members of ATP-binding cassette (ABC) transport superfamily (Table 29.5) (see also Chapter 11) [47]. They are concerned with the secretion of bile, but are important also in almost every cell organelle. The spectrum of disease caused by these defects is therefore diverse. Although usually familial with autosomal recessive inheritance, sporadic forms are identified.
Table 29.5. Genetic cholestatic syndromes
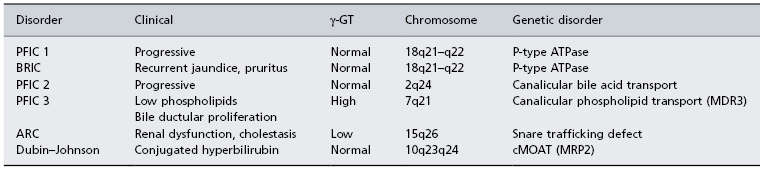
PFIC, progressive familial intrahepatic cholestasis; BRIC, benign recurrent intrahepatic cholestasis; γ-GT, γ-glutamyl transferase; MDR3, multiple drug resistance 3; MRP2, multidrug resistance protein 2; ARC, arthrogryphosis, renal dysfunction and cholestasis; cMOAT, canalicular multispecific organic anion transporter.
Progressive Familial Intrahepatic Cholestasis Type 1
Progressive familial intrahepatic cholestasis type 1 (PFIC) was formerly called Byler’s disease because of the association with the large numbers of an Amish kindred affected in Pennsylvania. The defect is now described from many parts of the world, including the Netherlands, Sweden and in Arab populations. Inheritance is autosomal recessive. It presents with cholestasis which eventually progresses to biliary fibrosis and cirrhosis. Liver transplantation is usually necessary in the first decade of life. Characteristically, serum γ-GT is low whereas alkaline phosphatase and serum primary bile acids are increased. Biliary bile acids are reduced.
The genetic defect has been mapped to the FIC1 locus on chromosome 18q21–q22 to a region encoding a P-type ATPase (ATP8B1) involved in aminophospholipid transport between membrane leaflets [48]. There is defective bile acid transport at the canalicular membrane.
Liver transplantation is inevitable, but post-transplant diarrhoea is a significant problem.
Benign Recurrent Intrahepatic Cholestasis
This presents with recurrent episodes of jaundice and pruritus, particularly in girls on the contraceptive pill [49]. The serum γ-GT is not elevated. The gene defect has been mapped to the FIC1 locus, the same region as PFICI 18q21–q22.
Progressive Familial Intrahepatic Cholestasis Type 2
This cholestatic disease is due to a mutation on chromosome 2 [50]. The gene encodes the human bile salt export pump (BSEP, ABCB11), an ATP-binding cassette transporter [51]. It does not involve the PFICI locus but has been mapped to a locus on chromosome 2q24. This encodes the BSEP gene. The primary defect is a defective canalicular bile acid transport pump.
Patients present with cholestasis, giant cell hepatitis with fat malabsorption and pruritus. Serum γ-GT is normal and bile ductular proliferation is not seen. The disease has a variable progression to cirrhosis and liver transplantation.
Progressive Familial Intrahepatic Cholestasis Type 3
This form of neonatal cholestasis is marked by an elevated serum γ-GT and bile ductular proliferation. There is a mutation in the multiple drug-resistant gene (MDR3 or P glycoprotein 3) on chromosome 7q21 (Table 29.5). This moves phospholipid from the inner leaflet of the canalicular membrane to the outer leaflet, which faces the canalicular lumen [52]. Serum phospholipid is low. Bile acid transport is unimpaired as is bile flow, but in the absence of phospholipid, the bile acids prove toxic to cholangiocytes and hepatocytes.
Treatment and Outcome
Routine interventions for chronic cholestatic liver disease are required for all three forms. Treatment with ursodeoxycholic acid (20 mg/kg per day) may be helpful. Partial biliary diversion surgery is sometimes used successfully for PFIC 1 and 2 [53]. Although variability in the disease course is evident, liver damage usually progresses and many require liver transplantation during childhood. Early development of hepatocellular carcinoma in the first 5 years of life has been reported with PFIC 2 [54].
Abnormal Bile Acid Synthesis
Defects in the synthesis of primary bile acids can cause decreased bile flow and abnormal transport and so cholestasis.
Bile acid synthesis defects may resemble PFIC type 2. 3β-hydroxy-C27-steroid dehydrogenase-isomerase deficiency results in cholestasis without pruritus and with normal serum γ-GT and bile acids. Cholestasis has also resulted from Δ4-3-oxosteroid-5β-reductase deficiency [55].
Coprostanic acidaemia results from a defect in the conversion of coprostanic to varinic acid. It is associated with progressive cholestasis and death by 2 years of age.
Zellweger’s cerebrohepatorenal syndrome is a fatal autosomal recessive condition with severe cholestasis. It is probably related to defective peroxisomal β-oxidation.
Treatment.
Toxic intermediates are formed which cause cholestasis by interacting with hepatic bile acid transport. Replacement of exogenous bile acids results in the generation of bile acid-dependent flow and a decrease in the synthesis of toxic bile salts. The administration of chenodeoxycholic acid, ursodeoxycholic acid and cholic acid [56,57] effectively reduce pruritus and transaminases and serum bilirubin levels fall.
Dubin–Johnson Syndrome (See Also Chapter 11)
This is not a cholestatic disease, but is marked by a rise in serum conjugated bilirubin. It is caused by a mutation of an ABC transporter, canalicular multispecific organic anion transporter (cMOAT) [58].
Symptomatic Treatment of Cholestatic Syndromes
The mainstay of treatment is intensive nutrition with adequate fat-soluble vitamin supplementation. Nutritional support is given by an increased energy intake of 120–150% of the estimated average, composed of 30–50% medium-chain triglyceride and high carbohydrate supplements. In severely malnourished infants, over-night or continuous nasogastric feeding may be required.
Fat-soluble vitamins are replaced orally by vitamin A 5000–15000 IU/day, vitamin D (α-calcidol) 50 ng/kg per day, vitamin K 2.5–5 mg/day and vitamin E 50–200 mg/day.
Pruritus is managed by a combination of phenobarbitone, ursodeoxycholic acid (10–20 mg/kg body weight), rifampicin (3 mg/kg) or by cholestyramine flavoured with apple purée, tomato juice or chocolate syrup [59].
Other Causes of Cholestatic Jaundice
Neonatal Lupus Erythematosus Syndrome
This presents as neonatal cholestasis and hepatitis [60]. Cutaneous lupus erythematosus and congenital heart block are associated.
Arthrogryphosis, Cholestasis, Renal Tubular Dysfunction (ARC)
This rare autosomal disorder presents with cholestasis, biliary hypoplasia, renal dysfunction and a low γ-GT. It is usually fatal and infants die of liver and renal failure within 12 months. The genetic defect is now known to be a disorder in a snare protein (VPS33B) involved in the regulation of vesicle and membrane fusion. It is the first human disorder associated with mutations in a gene involved in regulation of the SNARE-mediated mechanism of membrane tether and fusion and may lead to further understanding of the pathophysiology of biliary disorders [61].
Neonatal Sclerosing Cholangitis
This presents in early infancy with conjugated hyperbilirubinaemia which then resolves.
The aetiology is unknown but it may have a genetic basis [62]. The clinical picture includes: jaundice, development of hepatosplenomegaly, biliary cirrhosis and portal hypertension.
Laboratory investigations indicate elevated serum alkaline phosphatase and γ-GT. Endoscopic or percutaneous cholangiography demonstrates beaded irregularity of medium to large intrahepatic bile ducts in all patients and in extrahepatic ducts in 80%. Liver histology shows portal fibrosis with ductal proliferation developing into biliary cirrhosis. Surgical treatment is not indicated, but supportive management with nutrition is required. Most children require liver transplantation at some stage.
Structural Abnormalities: Biliary Atresia and Choledochal Cyst
The commonest causes of extrahepatic or structural biliary obstruction in neonates are biliary atresia or choledochal cysts.
Biliary Atresia
Extra hepatic biliary atresia is a disease of unknown aetiology with no proven genetic basis [63]. It is a rare disease which occurs with the frequency of approximately 1 : 15 000 live births [64,65]. The disease affects both intra- and extrahepatic ducts with progressive destruction leading to cholestasis, fibrosis and cirrhosis. The disease is classified according to the extent of biliary damage. Type I biliary atresia affects the common bile duct with a cystic proximal bile duct; type II affects the common hepatic duct; type III, which is the most common, affects the whole of the extrahepatic biliary tree.
There appear to be two clinical phenotypes. There is a syndromic or embryonic form that accounts for 10–20% of cases and is distinguished by other congenital anomalies such as polysplenia, situs inversus, cardiac anomalies (e.g. atrial and ventricular septal defects), absence of the inferior cava etc. (biliary atresia splenic malformation syndrome) [66]. The perinatal or acquired form is more common and represents 80–90% of cases.
Developmental Aspects
The biliary passages may fail to develop from the primitive foregut bud. The gallbladder may be absent or the biliary tract represented only by a gallbladder connecting directly with the duodenum. The more usual defect is failure of vacuolation of the solid biliary bud. This is usually partial and rarely extends throughout the biliary tree.
Pathogenesis
The underlying pathogenesis is unknown, but is likely to be multifactorial based on the interaction of genetic and environmental factors. Research to date has focussed on defects in morphogenesis, immunological dysregulation, viral infection such as reovirus or cytomegalovirus, or toxin exposure [67].
Pathology
The ducts may be absent or replaced by fibrous strands. Bile is absent from the extrahepatic biliary system including the gallbladder.
Clinical Features
There are more females than males, and all races are affected. Infants are full term with a normal birth weight and present with persisting jaundice from the second day of life. Other features include pale stools, dark urine, failure to thrive and hepatomegaly. Splenomegaly may be found but is a late feature and implies significant hepatic fibrosis and early cirrhosis [68]. Occasionally, infants present with bleeding from vitamin K-responsive coagulopathy, which is more common in breast-fed infants who did not receive vitamin K perinatally. About 5% of all infants will have had an abnormal antenatal maternal ultrasound. This is due to the presence of a cyst within the otherwise obliterated biliary tree and is detectable from around 22 weeks of gestation.
The diagnosis is suggested by a serum conjugated bilirubin exceeding 100 µmol/L (5 mg/dL), a serum alkaline phosphatase exceeding 600 IU/L, γ-GT exceeding 100 IU/L. ALT and AST are between 100 and 200 IU/L. Fasting ultrasound shows an absent or contracted gallbladder. Biliary scintigraphy with 99mTc-TEBIDA shows no excretion of isotope from liver into bowel 24 h after administration. Liver histology shows fibrosis, cholestasis and proliferation of biliary ductules with a variable number of giant cells. There is paucity of interlobular ducts.
The diagnosis is usually confirmed at laparotomy with or without cholangiography in which the atretic biliary tree is shown. It is usual to carry out palliative surgery, the Kasai portoenterostomy. Some infants who present late (>100 days) with established cirrhosis may be candidates for liver transplantation as a primary procedure. The natural history of this disease without medical or surgical management treatment is that the majority of children die within 2 years of birth from end-stage liver failure.
Surgical Management
The Kasai portoenterostomy is based on excision of the entire obliterated biliary tree to expose communicating biliary channels and create a Roux loop from the proximal jejunum which is anastomosed to the cut liver surface [69]. Restoration of biliary flow is considered the measure of success but depends on the age of surgery, the expertise of the surgeon and the extent of fibrosis at operation [65,70]. It is defined as normalization of serum bilirubin within 6 months of the procedure and in a recent UK report this was achieved in 57% of cases [71].
Age at Surgery.
Many studies have shown an improved outcome in terms of clearance of jaundice and native liver survival, the earlier the portoenterostomy is performed. Although the age at operation is an important factor, it may be a proxy for the extent of liver damage. Although initial results suggested that children operated on at 40 days of age did not show a significant survival advantage [72], recent data from Japan [73,74] suggest a clear advantage for those infants operated on earlier than 30 days of age, no difference in outcome in those operated on between 30 and 90 days, and a significant disadvantage only for those operated on later than 90 days.
Surgical Expertise.
The most important variable is the experience of the surgeon and the surgical centre. Recent studies have clearly demonstrated that the short and long-term outcome for children with biliary atresia is closely related to the experience of the surgeon and the centre [65,74,75]. These data led to the UK Department of Health directive to centralize surgery for all infants with suspected biliary atresia, which has been shown to improve the outcome [71].
Histology.
Although it has been suggested that the extent of histological abnormality at operation [76] or the morphology of the extra hepatic biliary remnant influence both short and long-term outcome, this has not been found consistently [77].
Medical Management
The important aspects of postsurgical management include prevention of complications, provision of nutritional and family support, requiring a multidisciplinary approach. This includes prevention of cholangitis, initially with intravenous antibiotics and subsequently with prophylactic low-dose oral antibiotics, which can be rotated at 8 or 12-week intervals (e.g. amoxycillin 125 mg/day, cephalexin 125 mg/day, trimethoprim 120 mg/day) for 12 months as a minimum. Ursodeoxycholic acid (20 mg/kg per day) may also be effective in encouraging bile flow and bile drainage. Nutritional support is essential to prevent malnutrition, overcome fat malabsorption and reduce the effect of excess catabolism. Fat-soluble vitamin supplementation should include vitamin A 5000–15 000 IU/day, vitamin D (alfacalcidol) 50 ng/kg per day, vitamin E 50–200 mg/day and vitamin K 2.5–5 mg/day.
The role of corticosteroids in improving biliary drainage is controversial. A number of small retrospective studies have suggested a beneficial effect with improved bile drainage and survival with native liver in children [69] but a prospective randomized placebo-controlled study in 71 infants found no significant effect on either native liver survival or proportion who cleared their jaundice [69].
Prognosis
Immediate complications include technical problems such as biliary leaks, exacerbation of ascites and ascending cholangitis. Longer-term complications include fat malabsorption and malnutrition, which leads to fat-soluble vitamin deficiency.
As the disease is progressive, all children will develop portal fibrosis, cirrhosis and portal hypertension, even if bile drainage has been established, which is more likely if there is recurrent cholangitis. However, 80% of children who have had a successful portoenterostomy are likely to survive more than 10 years with their native liver and achieve good quality of life [69,78]. Biliary atresia is the commonest indication for liver transplant in children. Currently, 76% of children under the age of 2 years who require transplantation have been born with biliary atresia (European Liver Transplant Registry 2005). The 15-year post-transplant survival is more than 80% [79].
Choledochal Cyst (See Also Chapter 14)
Choledochal cyst is a congenital malformation of the biliary system which may be identified in the fetus by antenatal ultrasound [80,81].
Clinical Features and Diagnosis
Choledochal cysts present with jaundice, abdominal mass and pain with acholic stools, but this is an unusual presentation in the neonatal period. There is a female predominance (female : male is 5 : 1). The diagnosis is made by identifying the choledochal cyst by ultrasound examination of the liver and confirmed by cholangiography, either percutaneous or endoscopic. Liver function tests are consistent with biliary obstruction and liver biopsy may demonstrate large bile duct obstruction and fibrosis, which is reversible after surgery.
Surgical treatment includes excision of all the affected ducts and re-establishment of biliary drainage by forming a hepaticojejunostomy. Drainage of the cyst into adjacent duodenum or jejunum is now contraindicated, because of the potential malignant transformation. The results of surgery are excellent, cholangitis is an occasional complication, and there is a 2.5% risk of malignancy in the residual biliary tree in adult life [82].
Spontaneous Perforation of the Bile Ducts
This is a rare complication in which perforation occurs at the junction of the cystic and common hepatic ducts, perhaps due to a congenital weakness, inspissated bile or gallstones. Infants may present at any age from 2–24 weeks of age with abdominal distension, ascites, jaundice and acholic stools. Biliary peritonitis, with bile in hydroceles, hernial sacs and umbilicus may be obvious.
The diagnosis is confirmed by abdominal ultrasound, which may show free intraperitoneal fluid and dilated intrahepatic ducts. Biochemical liver function tests may be abnormal, with conjugated hyperbilirubinaemia and raised alkaline phosphatase and γ-GT. If the biliary leak is large, liver function tests may be virtually normal. Cholangiography shows the blocked cystic duct with the hepatic duct leak. Hepatobiliary scanning will demonstrate isotope into the peritoneal cavity. Treatment includes peritoneal drainage followed by repair of the perforation. The results of surgery are good.
Inspissated Bile Syndrome and Cholelithiasis
Bile duct obstruction secondary to inspissated bile syndrome may be secondary to total parenteral nutrition, prolonged haemolysis and dehydration. It is more common in premature babies or those undergoing major surgery. The clinical picture is of biliary obstruction with pale stools, dark urine and abnormal liver function tests. The diagnosis is made by ultrasound, which demonstrates a dilated intra- and extrahepatic duct system with biliary sludge. Percutaneous transhepatic cholangiography will outline the anatomy and may be therapeutic with lavage of the biliary tree, but laparatomy and decompression of the biliary tree may be required. The use of ursodeoxycholic acid (20 mg/kg) and cholecystokinin may prevent the need for either surgical or radiological intervention.
Cholecystitis may occur in infants in association with gallstones from haemolysis or total parenteral nutrition, while acalculus cholecystitis may occur as part of generalized sepsis. Operative cholecystectomy (rather than laparoscopic) is the treatment of choice in this young age group for symptomatic cholecystitis in association with gallstones [83].
In older children, cholecystitis and gallstones may be associated with blood dyscrasias or congenital anomalies of the biliary tract, such as choledochal cysts or biliary atresia.
Acute Liver Failure in Infancy
Acute liver failure in infancy usually presents with multisystem involvement. The diagnosis may initially be difficult as jaundice may be a late feature. Infants are usually small for gestational dates, with hypotonia, severe coagulopathy and encephalopathy. Neurological problems, such as nystagmus and convulsions, may be secondary to cerebral disease or encephalopathy. Renal tubular acidosis is common. Investigations include a search for multiorgan disease.
Galactosaemia
This rare autosomal disorder is secondary to a deficiency of galactose-1-phosphase uridyltransferase (GALT), essential for galactose metabolism, in the liver and red blood cells. It is inherited as an autosomal recessive with a frequency of between 1 in 10 000 and 1 in 60 000. There are more than 150 mutations reported in the GALT gene [84], and this genetic heterogeneity contributes to the wide phenotypic heterogeneity. A significant reduction of the transferase is found in heterozygotes.
Clinical Picture
Acute illness results from the accumulation of the substrate galactose-1-phosphate (gal-1-P) following the introduction of milk feeds [85]. Infants present with collapse, sepsis, hypoglycaemia, and encephalopathy in the first few days of life or with progressive jaundice and liver failure. Ascites and hepatosplenomegaly may be noted. Cataracts are present. The disease may be complicated by Gram-negative sepsis, which stimulates a life-threatening severe bleeding diathesis.
The condition should be considered in all young patients with cirrhosis and even in the adult if there are suggestive features such as cataract.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


