The other key feature to make the diagnosis of NAFLD is the exclusion of ethanol exposure as a significant factor. This remains controversial, with little agreement between studies as to whether this means total abstinence or consumption below a threshold level. There are studies showing less or no apparent additional histological injury (and perhaps less injury) with modest alcohol consumption in NASH [13], and others showing obesity as a risk factor for alcohol-induced liver disease [14]. In general, daily consumption of less than 20 g in women and 30 g in men results in a low risk for the development of alcoholic liver injury, although this leaves a grey area for those with less modest daily consumption, but still below the ranges associated with alcohol-induced liver disease. Lifetime ethanol exposure rather than daily consumption suggest that about 10% of patients diagnosed with NASH may have a component of alcohol-related steatohepatitis (ASH) [15].
Liver Biopsy, Classification of Non-Alcoholic Fatty Liver Disease and Non-Invasive Markers of Non-Alcoholic Steatohepatitis
Although non-invasive markers of histological injury are under study, the accurate diagnosis of NASH remains dependent on specific histological parameters in a biopsy. The key parameters include steatosis (usually mixed macro- and microvesicular), cellular ballooning, inflammation and fibrosis which ranges from slight perisinusoidal fibrosis to bridging and cirrhosis [16–18]. The major indicators of injury have been incorporated into a score commonly called the NAS (NAFLD Activity Score) and staging system of fibrosis, which currently is being used in most clinical trials [19]. These features have also been used to divide NAFLD into four types: (1) simple steatosis, (2) steatosis with inflammation alone, (3) steatosis with inflammation and ballooning, and (4) steatosis with inflammation and fibrosis [8]. In general, the latter two types constitute NASH (because ballooning and fibrosis usually parallel each other and are typically associated with inflammatory infiltrates). The first two types can be grouped into ‘NNFL’ (non-NASH fatty liver) [20]. The division of NAFLD into NASH and NNFL is significant because of their relationship to prognosis and appropriate therapy (Fig. 28.2 and Table 28.1).
Table 28.1. Working classification of non-alcoholic fatty liver disease
| NNFL |
| Type 1 NAFLD: Steatosis with no inflammation or fibrosis |
| Type 2 NAFLD: Steatosis with non-specific lobular inflammation but absent of fibrosis or hepatocyte ballooning |
| NASH |
| Type 3 NAFLD: Steatosis with inflammation and fibrosis of variable levels (NASH) |
| Type 4 NAFLD: Steatosis, inflammation, hepatocyte ballooning, and fibrosis or Mallory–Denk bodies (NASH) |
NNFL refers to non-NASH fatty liver and indicates the development of steatosis in the absence of significant ethanol exposure and without evidence of significant cell injury. Synonyms include simple steatosis and NAFL (non-alcoholic fatty liver). NASH, non-alcoholic steatohepatitis; NAFLD, non-alcoholic fatty liver disease.
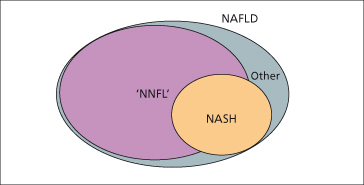
Fig. 28.2. Non-alcoholic fatty liver disease (NAFLD) exists as a spectrum that includes non-alcoholic steatohepatitis (NASH), which constitutes about one-third of cases. The remainder of the cases consist mostly of non-NASH fatty liver termed ‘NNFL’, and a small representation of other conditions such as storage diseases, discussed in the final section of this chapter.
Although liver biopsy remains the ‘gold standard’, there are practical limitations, including costs and risks. Importantly, sampling error is well recognized, and longer cores are needed for accurate fibrosis staging [21]. Minimal sizes are debated but most recommend 2 cm or more. Non-invasive alternatives, which include a number of clinical scoring systems, as well as laboratory tests, cannot distinguish intermediate levels of injury [22]. However, these markers may eventually provide an accurate means of distinguishing NASH and NNFL, grading the severity of NASH and monitoring the response to therapeutic interventions. Promising parameters include collagen-related metabolites as indicators of fibrosis, adipocytokines as indicators of systemic fat metabolism and insulin signalling and cytokeratin 18 fragments as a measure of apoptosis pathways activation [23,24].
Clinical Features
Most patients with NASH have insulin resistance and the metabolic syndrome (Table 28.2) with central or visceral obesity [25–28]. Insulin resistance can be demonstrated in the liver (failure to suppress glucose output), in skeletal muscle (diminished glucose uptake) and in adipose tissue (failure to suppress lipolysis and release of fatty acids). Indeed, resistance is likely to result from systemic lipotoxicity (Table 28.3) [29,30]. Hypertension, hyperlipidaemia and type 2 diabetes, components of the metabolic syndrome, can cause confusion regarding possible drug-induced liver enzyme abnormalities in patients using antihypertensives, antidiabetic and antihyperlipidaemic agents, who are actually suffering from NASH.
Table 28.2. National Cholesterol Education Program: Adult Treatment Program III (NCEP ATP-III) Guidelines—metabolic syndrome components
| Risk factor | Defining level |
| Abdominal obesity | Waist circumference |
| Men | >102 cm (>40 in) |
| Women | >88 cm (>35 in) |
| Triglycerides | ≥150 mg/dL |
| HDL cholesterol | |
| Men | <40 mg/dL |
| Women | <50 mg/dL |
| Blood pressure | ≥130/ ≥85 mmHg |
| Fasting blood glucose | ≥110 mg/dL |
Reaching the defining level for any three of the parameters satisfies the clinically applicable definition of metabolic syndrome.
Table 28.3. Key sites of insulin resistance
| Tissue | Dysfunction |
| Adipose | Failure to suppress hormone-sensitive lipase activates release of FFA from triglyceride stores |
| Liver | Failure to suppress glucose production/ release from glycogenolysis and gluconeogenesis |
| Muscle | Failure in glucose uptake due to: decreased translocation of GLUT-4 transporter increased myocyte lipid stores impaired mitochondrial function |
FFA, free fatty acids; GLUT-4 transporter, transmembrane insulin-responsive transporter found in adipose and muscle primarily responsible for glucose uptake from the circulation/ periphery.
Most patients also have impaired exercise tolerance (measured by oxygen consumption during graded exercise). These findings are consistent with ‘metabolic obesity’ even in those with relatively low body mass index (BMI) [31]. The average age for NASH patients is 40–50 years and for NASH-related cirrhosis it is 50–60 years. However, the emerging obesity epidemic has resulted in increasing numbers of children with this disease—sometimes with advanced fibrosis [32]. About 20% of patients report a family history of unexplained liver disease (see below). Other common associations include polycystic ovary syndrome, sleep apnoea and small bowel bacterial overgrowth [33,34]. The latter may explain the frequent association of vague abdominal symptoms seen in these patients. Many of these findings, including hepatic dysfunction, can be unified under the concept of systemic lipotoxicity, which implicates toxicity related to excessive intracellular fatty acid accumulation which, in turn, alters cellular metabolism in many tissue types [35]. The occasional description of unexplained neurodegenerative disease in association with NASH also points towards the systemic nature of this disease [36]. The most common presentation of NAFLD is the detection of mildly abnormal aminotransferases during a routine clinical evaluation. However, there is a tendency for these levels to decline with progressive disease in parallel with decreasing steatosis, as fibrosis worsens. Thus it is important to examine the patient closely for stigmata of cirrhosis, including a firm, palpable liver and cutaneous signs such as palmer erythema and spider angiomata, as well as laboratory signs such as thrombocytopenia. Because NASH is often clinically silent during the early stages, it is not uncommon to make a diagnosis of cirrhosis ‘by chance’, for example during gall bladder surgery. Patients may also present with complications of portal hypertension, and occasionally with acutely decompensated, but previously unrecognized, disease [37]. Gastrointestinal bleeding from GAVE (gastric antral vascular ectasia) is sometimes seen.
Other physical findings include acanthosis nigricans (pigmentation and skin thickening in the axillae and posterior neck), which is more common in children. A prominent dorsal fat pad (buffalo hump) is common and has been associated with more severe histological disease [38]. Evidence of lipodystrophy should also be sought (see below).
Laboratory Testing
Elevations of serum AST (aspartate aminotransferase) and ALT (alanine aminotransferase) are usually less than two times normal [39]. However, concentrations within the reference range may be associated with significant disease [40]. Aminotransferase patterns may be helpful in staging NASH, as an AST : ALT ratio greater than 1 suggests progression to more advanced fibrosis [41]. However, this pattern appears to be less reliable in patients on thiazolidinediones or statin medications. Serum IgA levels may be mildly elevated, possibly as a result of oxidative injury and the formation of neoantigens with a biliary mucosal B cell response, as previously suggested in alcohol-related liver disease [42,43]. Hyperuricaemia is common and thought to result from abnormalities in ATP metabolism resulting in ADP accumulation and excessive purine disposal. Antinuclear antibodies are detected in 25–30% of NASH patients. The mechanism for their development is not well understood. Disorders of the thyroid have also been associated with NAFLD.
Other laboratory abnormalities include abnormal lipoprotein profiles and markers of insulin resistance or diabetes. There are no consistent patterns of dyslipidaemia although hypertriglyceridaemia is usually found. Insulin resistance can be measured by the QUICKI test (quantitative insulin sensitivity check index) or the HOMA test (homeostasis model assessment) both of which are derived from the euglycaemic hyperinsulinaemic clamp test with mathematical modelling of fasting insulin and glucose levels. A modification of the QUICKI test can also be used as a measure of insulin resistance in adipose tissue.
Mitochondriopathies and Lipodystrophy
Mitochondrial abnormalities similar to those seen in primary mitochondrial diseases have been observed in NASH [30,44]. The presence of ophthalmoplegia, neurodegenerative diseases, retinopathy, neural deafness or severe lipomatosis should raise suspicion of a primary disorder. Insulin resistance and dyslipidaemia are found in conditions resulting from mitochondrial DNA mutations such as symmetrical lipomatosis or Madelung’s disease and the MIDD syndrome (maternally inherited diabetes and deafness) [45,46]. The lipodystrophies are linked to insulin resistance and to NASH [47]. Focal forms of these disorders are known, but how often these occur among otherwise typical NASH patients is not established—marked sparing of the limbs in an obese individual should raise suspicion.
Epidemiology of Non-Alcoholic Fatty Liver Disease
The prevalence of NAFLD is remarkably high in populations of both industrialized and developing countries, although there is variation depending on the criteria used and the population studied [20,48]. In one study of adults based on histological findings, mild to severe steatosis was shown in 70% of obese patients compared to 35% of lean patients. Steatohepatitis was found in 18.5% of obese patients, compared to 2.7% of lean patients [49]. A relatively high prevalence has also been observed in countries with typically lower BMI such as in regions of Asia [50,51]. The annual incidence of NAFLD in prospectively followed adult populations is estimated to be about 3–5% [48]. NAFLD is also well documented in children. A US autopsy study of 742 children resulted in prevalence of 9.6% [52], and was more common in boys than girls (2 : 1 ratio). Other non-histology-based population studies in the USA, Europe and Asia have shown the prevalence in children to be approximately 2–3% [53–56]. Consistent with a strong familial component, there is a high prevalence of NAFLD among the adult relatives of affected children [57].
In the primary care setting, NAFLD accounts for at least one-third of cases of suspected chronic liver disease [58]. Among patients with abnormal liver enzymes, NAFLD accounts for 40–80% of cases, with its prevalence strongly influenced by the presence of coexisting obesity, diabetes and dyslipidaemia [59]. In severely obese patients (usually defined as BMI >35 kg/m2), the prevalence of steatosis is over 90% from series of patients undergoing bariatric surgery [60]. About one-third of these patients have NASH, two-thirds have NNFL and 2–3% of patients have NASH-related cirrhosis. In another study, NAFLD was evident in 94% of obese patients (BMI ≥30 kg/m2), 67% of overweight patients (BMI ≥25 kg/m2), and 25% of normal-weight individuals [61].
From another perspective, three-quarters of type 2 diabetic patients have steatosis. The coexistence of diabetes in NAFLD patients more than doubles the prevalence of cirrhosis from 10 to 25% [62,63]. Among patients with hyperlipidaemia, at least two-thirds with hypertriglyceridaemia and one-third with hypercholesterolaemia have fatty liver by ultrasound imaging [64]. Refinement of the epidemiology of NAFLD in different forms of hyperlipidaemia is needed.
Ethnic Variation in Non-Alcoholic Fatty Liver Disease
The relationships between obesity, diabetes, hyperlipidaemia and NAFLD are influenced by ethnicity. People of African-American descent have significantly less hepatic steatosis in spite of a relatively high prevalence of obesity and diabetes [65]. In contrast, people of Hispanic-American descent have a higher prevalence, while those of primarily northern European and Asian-American descent have an intermediate prevalence of steatosis [66,67]. Although there is a constant relationship between liver triglyceride content and intraperitoneal fat (visceral adiposity) within ethnic groups, there is a substantial degree of dissociation between insulin resistance and both steatosis and visceral adiposity in some ethnic groups [68]. This unusual situation suggests the development of different forms of the metabolic syndrome based on the hypothesis of the ‘thrifty genome’ in ancient human evolution [69,70]. A possible genetic basis for ethnic variation has been identified based on genetic polymorphisms of a liver-expressed transmembrane phospholipase, PNPLA3 (patatin-like phospholipase domain-containing protein 3) [71].
Familial Associations
Familial clustering of NASH and NAFLD could represent inherited genetic predisposition or common environmental factors such as dietary habits or activity levels [72–74]. The finding of impaired skeletal muscle mitochondrial metabolism and insulin resistance in the offspring of patients with type 2 diabetes suggests a genetic risk related to intracellular fat metabolism [75]. In a study of fatty liver by H1 MR spectroscopy among siblings and parents of overweight probands with and without fatty liver, steatosis was detected in 17% of siblings and 37% of parents of the overweight group without fatty liver compared to 59% and 78% of siblings and parents respectively in the overweight group with fatty liver [52]. Taken together with studies of ethnic variation, these findings suggest a strong genetic component in the development of both NAFLD in general, and NASH in particular, although it remains to be seen whether the pattern of severity also follows predictable patterns.
Pathogenesis of Non-Alcoholic Fatty Liver Disease and Non-Alcoholic Steatohepatitis
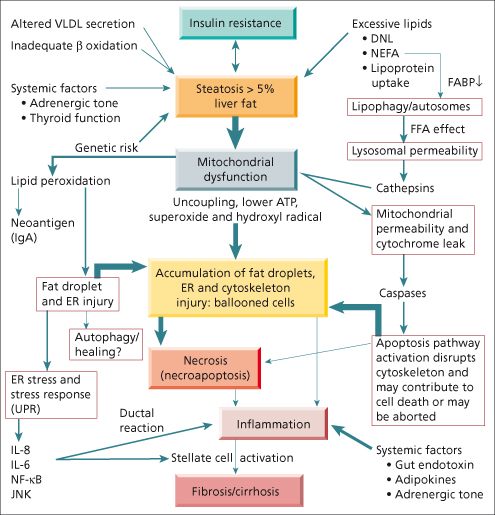
Although the mechanism of hepatocellular injury in NASH can be encapsulated in the ‘two hit’ hypothesis [76]—accumulation of fat followed by oxidative injury—it is increasingly evident that the mechanism involves a complex cascade of events leading to hepatocellular injury, most evident as cellular ballooning and cell death (Fig. 28.3). Studies of the ballooned cell from various perspectives lead to an emerging concept of ‘multiorganelle’ failure, with impairment of critical organelles and inadequate compensatory pathways. These pathways are both driven by and contribute to systemic abnormalities, especially related to insulin resistance and disturbed energy homeostasis.
Fig. 28.3. Summary of the mechanism of non-alcoholic steatohepatitis (NASH) pathogenesis. VLDL, very low density lipoprotein; β oxidation, mitochondrial oxidation of fatty acids; DNL, de novo lipogenesis from carbohydrates such as glucose; NEFA, non-esterified fatty acids resulting from unrestrained lipolysis in adipose tissue. FABP is fatty acid binding protein, which is diminished in NASH (see text). FFA is free fatty acids, an excess appears to promote permeabilization of lysosomes and mitochondria with release of cathepsins and cytochrome C, inducing caspases which activate apoptosis pathways. ER is endoplasmic reticulum the dilation of which, along with accumulation of small fat droplets, contribute to cellular ballooning. UPR is the ‘unfolded protein response’ which defines a certain form of endoplasmic reticulum dysfunction. IL-8 and IL-6 refer to interleukins 8 and 6, which are increased in concert with JNK (c-Jun N-terminal kinase), and NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) in the setting of ER dysfunction (see text). Ductal reaction refers to the activation of progenitor cells near the portal zones, which are likely to contribute to both portal inflammation and fibrosis. Stellate cells are the major collagen producing cells of the liver, which lead to fibrosis and eventually to cirrhosis.
Mechanisms of Steatosis
Lipid accumulation in the liver results from an imbalance between overall calorie intake and systemic calorie utilization characteristic of the metabolic syndrome. Hepatic fat results from several possible mechanisms including synthesis of new fatty acids, especially from carbohydrate precursors (de novo lipogenesis), uptake of circulating free fatty acids (non-esterified fatty acids, NEFA) derived from adipose tissue lipolysis, uptake of diet-derived chylomicron remnants or uptake of very low density lipoprotein (VLDL)-derived low density lipoprotein (LDL) remnants. Liver fat can be disposed of by either oxidation or lipoprotein secretion especially as VLDL. NAFLD appears to be driven especially by NEFA uptake, de novo lipogenesis and altered lipid export. Recycling of lipids through the mechanisms of autophagy is an emerging aspect of pathogenesis, discussed below.
Regulation of Lipid Synthesis
Within the hepatocyte, lipid stores are primarily regulated by two main transcription factors: sterol regulatory element binding protein (SREBP), governed by insulin and dietary fatty acids, and carbohydrate response element binding protein (CREBP), governed by ambient glucose levels [77–79]. SREBP and CREBP stimulate nuclear transcription of the enzymes responsible for fatty acid synthesis, and subsequently their esterification into triglyceride, stored as triacylglycerides within cytosolic fat droplets, or exported as VLDL.
Biochemistry of De Novo Lipogenesis
At the molecular level, the synthesis of 16 carbon unsaturated palmitic acid (the initial end point of de novo lipogenesis), begins with translocation of carbohydrate-derived acetyl-CoA subunits as citrate, which pass through the mitochondrial membrane to the cytosol. ATP-dependent cytosolic condensation of acetyl-CoA subunits into palmitate depends on the activity of a key enzyme, acetyl CoA carboxylase, which is regulated by adrenaline (epinephrine), glucagon and insulin, and which activates formation of malonyl CoA from acetyl-CoA. Molecules of malonyl CoA then serve as the building blocks for assembly of the 16-carbon palmitic acid fatty acid, through a series of condensations catalysed by fatty acid synthase. Malonyl CoA also inhibits mitochondrial β-oxidation of fatty acids by blocking the carnitine shuttle, by which fatty acids destined for oxidation are moved into the mitochondrion.
Once formed, palmitate can undergo elongation in the endoplasmic reticulum to long-chain and very-long-chain fatty acids. Palmitate can also undergo desaturation and esterification to glycerol to form mono-, di- and triacylglycerols (triglycerides), which are incorporated into fat droplets in the endoplasmic reticulum or packaged through the activity of microsomal triglyceride transfer protein, in association with apolipoprotein B100 (apoB100), for secretion as VLDL [80].
Steatosis in Humans
Based on studies using radiolabelled precursors, 59% of triglyceride synthesis in human NAFLD results from uptake of adipose-derived NEFA while de novo lipogenesis (driven by SREBP and CREBP) accounts for about 26% and dietary sources for 15% [81]. The high burden of NEFA appears to derive predominantly from visceral fat, and represents failure of insulin to suppress the activity of hormone-sensitive lipase at adipose stores (see insulin resistance below). Incorporation of NEFA into triglycerides and their contribution to steatosis appears from experimental work to depend on the activity of acyl CoA: diacylglycerol acyltransferase 1 (Dgat1) [82]. However, the other sources of fatty acids are also significant. Compared to normal, de novo lipogenesis is increased from 5 to 15–25%, and spill-over from intestinal-derived chylomicrons is increasingly recognized, and may also be a source of additional oxidative stress [83–85].
Opposing the accumulation of liver fat, VLDL secretion is increased in NAFLD but plateaus at a hepatic triglyceride content of 10%, indicating limited compensation for high circulating NEFA [86]. Moreover, the secretion of apoB100, a key component of normal VLDL, is impaired in human NAFLD and may correlate with the secretion of a larger VLDL particle with greater triglyceride relative to its apoB100 content [28,87].
Mitochondrial Dysfunction
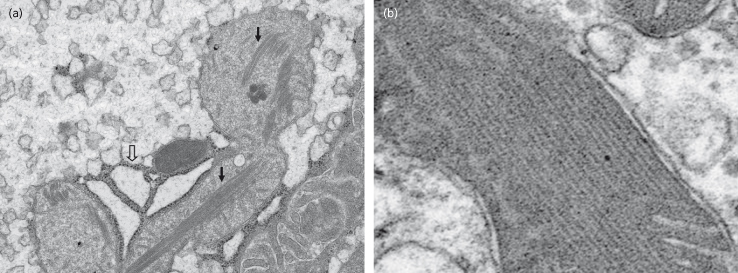
The accumulation of lipids in the liver is associated with an energy deficient state evidenced as diminished ATP content. This was shown experimentally over 50 years ago [88]. More recently, deficient hepatic ATP synthesis following intravenous fructose challenge in human NAFLD using 31P MR spectroscopy has been shown [89]. The mitochondria appear to be both a target and a source of pro-oxidant free radicals (superoxide and hydroxyl radicals) the effects of which are key distinguishing features of NASH, as opposed to NNFL [90,91]. Mitochondrial morphological changes are readily evident in human NASH and include swelling and intramitochondrial crystals (Fig. 28.4). The crystalline structures appear to be phase transitions of cristae phospholipid bilayers [92].
Fig. 28.4. Mitochondrial crystals in human non-alcoholic steatohepatitis (NASH). (a) Transmission electron microscopy of human NASH showing enlarged mitochondria containing intramitochondrial crystals (closed arrows) and dilated endoplasmic reticulum (open arrows). (b) Closer view of a mitochondrion containing intramitochondrial crystals (see text).
Impaired function of the mitochondrial electron transport chain has been observed in both animal and human studies. It is due in part to over-expression of uncoupling protein and to dysfunction of components of the electron transport chain [93,94]. Electron transport chain activity is reduced to 40–70% of normal in all of the major complexes (I–V) in human NASH [95]. Although impairment of oxidative phosphorylation is evident, it is uncertain whether or not, overall net fatty acid oxidation is decreased or increased—some studies document a surprising net increase in mitochondrial β-oxidation [30,96]. A clearer consensus exists regarding changes in mitochondrial permeability, leading to release of mitochondrial cytochrome c and apoptosis signalling, as discussed below [97]. Increased mitochondrial cholesterol has been proposed as a mechanism contributing to mitochondrial dysfunction and associated changes in permeability [98,99].
Lipid Composition in Non-Alcoholic Fatty Liver Disease
Fat composition studies in NAFLD showed that most of the stored lipid is composed of triglyceride, with a lesser component of free fatty acids, although both were significantly increased compared to normal subjects [100]. Both macro- and microvesicular fat droplets are present in NAFLD, although smaller droplets are often less evident by conventional light microscopy without either specific fat stains (such as oil red O) or by osmium fixed specimens for electron microscopy. Recent lipidomic analysis of liver tissue in human NAFLD have shown significant differences in NASH versus NNFL [101]. Stepwise increases in both triacylglycerol : diacylglycerol and free cholesterol : phosphotidylcholine ratios were noted from normal to NNFL to NASH subjects. Polyunsaturated fatty acids, such as eicosapentanoic acid and docosahexanoic acid, were relatively lower in NASH, leading to an elevated N6 : N3 ratio, suggesting a relative excess of proinflammatory N6 fatty acids such as arachidonic acid. Interestingly, the level of hepatic free fatty acids, although higher compared to controls, were not different in NASH versus NNFL—this is another unresolved point of controversy (see below). Increased ceramide, a potentially toxic intermediary in sphingolipid metabolism, in peripheral white adipose tissue has been detected in obese patients with fatty liver versus those without [102].
Lipid Peroxidation in Non-Alcoholic Steatohepatitis
Ample evidence in humans indicates that cellular injury in a lipid-loaded hepatocyte is initiated by impaired control of aerobic metabolism resulting in oxidative stress and lipid peroxidation [103–107]. Although cytochrome p450 (ω-oxidation) or peroxisomal fatty acid oxidation may contribute to free radical formation, the superoxide radical is primarily derived from mitochondria. Once formed, it is metabolized via superoxide dismutase to hydrogen peroxide. Hydroxyl radicals result from decay of hydrogen peroxide, in the presence of Fe2+ via the Fenton or Haber–Weiss reactions. Unless detoxified by glutathione, hydroxyl radicals damage other cellular constituents, including membrane fatty acids, proteins and DNA through direct binding [108]. Injury to the fatty acids produces lipid peroxidation—a branching, chain reaction stimulated by a free radical attack on unsaturated fatty acids which produces another free radical and a lipid hydroperoxide. The latter degrades in a reaction catalysed by iron to form a second lipid-based free radical thus amplifying the process [109]. Oxidative injury to the phospholipid monolayer of small fat droplets, which contain insulin-sensitive lipases (PAT family proteins), and to the endoplasmic reticulum may be particularly relevant to development of cellular ballooning, impaired disposal of toxic free fatty acids and hepatic insulin resistance [105,110,111]. Other by-products of oxidative injury include metabolites of nitric oxide, particularly in macrophages, and neoantigen formation, which may explain a link to serum IgA elevation discussed earlier.
Autophagy, Lysosomes, Fatty Acid Induced Injury and Apoptosis
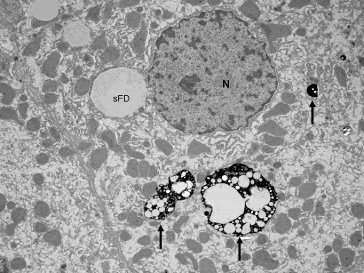
Disposal of accumulated and presumably injured fat droplets involves the process of lysosome-mediated autophagy (Fig. 28.5) [112,113]. Recent experimental work, mostly involving animal models or cell cultures, has indicated the relative stability of fat stored as triglycerides, compared to the potential toxicity of free fatty acids [114]. Impaired autophagy of small fat droplets, as well as diminished fatty acid binding protein shown in one human study, may contribute to accumulation of free fatty acids and cellular lipotoxicity [115]. Polymorphisms of the enzyme responsible for formation of triglycerides from diglycerides (Dgat or acyl-CoA: diacylglycerol acyltransferase) could also contribute to impaired disposal of free fatty acids [116]. Free fatty acids, in turn, alter lysosomal permeability leading to release of cathepsins (lysosomal proteases), which are associated with changes in mitochondrial permeability [117]. In cell culture models, this contributes to release of mitochondrial cytochrome C which activates caspases, leading to further activation of proapoptosis pathways, which are evident in human NASH [118,119]. Accumulation of palmitic acid has also been shown in cell culture to stimulate release of interleukin-8 from hepatocytes [120].
Fig. 28.5. Autophagosomes (arrows) containing ingested small fat droplets (sFD) in human NASH. N, nucleus.
Endoplasmic Reticulum Stress, Activation of Inflammation, Fibrosis and Cell Death
While final cell death may result from some combination of necrosis and apoptosis (necroapoptosis) [121], the activation of caspase 3 leads to fragmentation of cytokeratin 18. This is likely to contribute to formation of Mallory–Denk bodies, seen best in ballooned hepatocytes, and to cytokeratin 18 fragments detectable in blood [24,122]. Accumulation of free fatty acids and impaired function of endoplasmic reticulum (ER)-associated ApoB100 (an essential lipoprotein in ER-based VLDL synthesis) also contribute to ER stress with an accumulation of misfolded proteins within the ER [123,124]. ER stress, along with accumulation of free fatty acids and cell death, induce proinflammatory cytokines such as interleukin 8, through activation of transcription factors such as nuclear factor kappaB (NF-κB) and c-Jun N-terminal kinase (JNK) [120]. Importantly, this pathway appears to be active in human forms of the disease; the degree of activation of JNK related to ER stress distinguishes NASH from NNFL in human studies [125,126]. Modulation of this pathway also appears to improve antioxidant status in animal models [127]. Ultimately, activity in these pathways leads to accumulation of inflammatory infiltrates and activation of collagen-producing hepatic stellate cells (usually in close association with cellular ballooning) characterized by transition from a vitamin A rich quiescent cell to a proliferating myofibroblast [128]. Activation of collagen-producing stellate cells is mediated, at least in part, by activation of the Toll-like receptor [129]. Progression of fibrosis may also depend on an altered repair process with impaired hepatocyte replication and increased activity of hepatic progenitor cells leading to a ductular reaction in the portal tracts [130].
The Ballooned Cell
Morphologically, the ballooned hepatocyte, identifiable by deficiency of intact cytokeratin [131], is most clearly associated with active steatohepatitis in human NASH. It consists of an accumulation of multiple, small fat droplets in association with distorted mitochondria, dilated ER and Mallory–Denk bodies, supporting a close link between each of these processes (Figs 28.6, 28.7). Because multiple pathways are undergoing simultaneous failure, the process can be described as multiorganelle failure. In this setting, the final event is most likely to be necrosis, although activation of apoptotic pathways plays a substantial role. This course is the likely explanation for the characteristic appearance of histological necrosis concurrent with activation of apoptosis leading to necroapoptosis.
Fig. 28.6. Fat droplet accumulation and cytoskeletal injury in human non-alcoholic steatohepatitis (NASH). Serial images of (a) haematoxylin and eosin (H&E), (b) oil red O and (c) keratin 18 immunohistochemical (IHC) staining in human NASH. Arrows indicate ballooned cells detected by H&E and then by a fat stain using oil red O, showing accumulation of fat droplets, and then by IHC for cytokeratin 18 showing a deficiency of K18 and highlighting of a Mallory–Denk body (MDB; see text) indicating significant cytoskeletal injury. Lipid peroxidation is thought to underlie these processes.
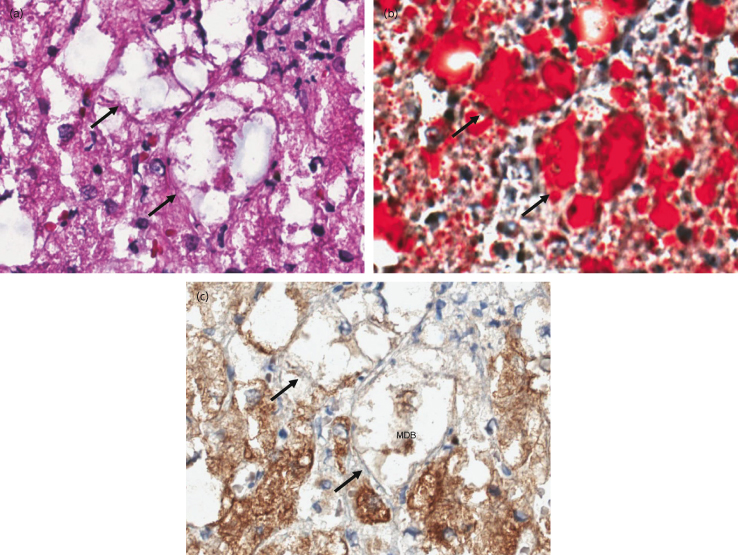
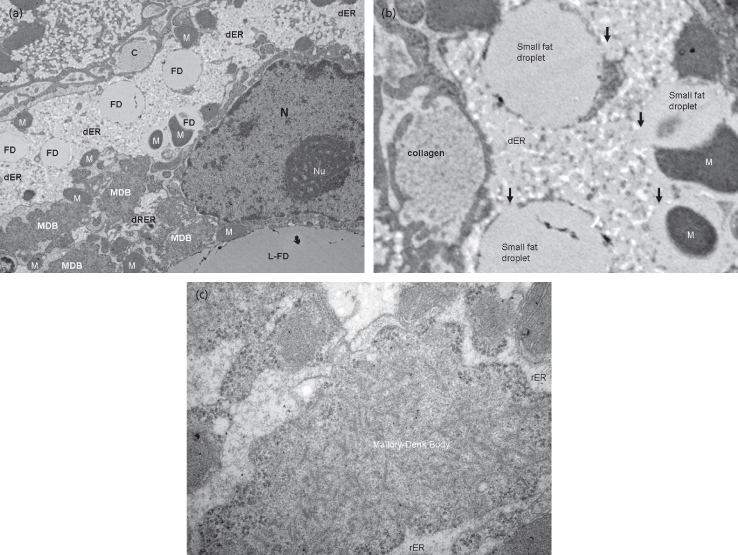
Fig. 28.7. Transmission electron microscopy in non-alcoholic steatohepatitis (NASH). (a) A closer view of a damaged hepatocyte shows: accumulation of fat droplets (FD) and a large fat droplet (L-FD); Mallory–Denk body (MDB), which represents collapse of the cytoskeleton; an extracellular bundle of collagen (C) as a marker of fibrosis; mitochondria (M) and dilated endoplasmic reticulum (dER) and rough endoplasmic reticulum (dRER). (b) Closer view of the fat droplets and associated endoplasmic reticulum dilation from the same sample. (c) Closer view of the MDB from the same sample.
Systemic Factors
Hepatic fat is regulated by other systems involved with energy homeostasis including insulin/glucagon, the adipose organ, the adrenergic system and the thyroid axis. The demonstration of cold-activated brown adipose tissue in healthy men and its inverse relationship to obesity emphasizes the integration of energy management and thermoregulatory systems [132]. Adipose-derived cytokines and adipocytokines (such as adiponectin) play a role through modulation of insulin activity and the inflammatory response to fatty acid-induced injury. Noradrenergic modulation of the inflammatory infiltrate has also been demonstrated in animal models [133]. In human NAFLD, hepatic and extrahepatic insulin resistance is present in the majority of patients and is virtually inseparable from the pathophysiology of the disease.
Insulin resistance is primarily mediated by excessive free fatty acids and occurs in multiple insulin end-organs including adipose, skeletal muscle and the liver (Table 28.3) [30,134,135]. At the molecular level, insulin resistance is characterized by a shift from tyrosine phosphorylation in the insulin receptor substrate to serine phosphorylation, which blunts the anabolic effects of insulin in multiple downstream pathways, including both metabolic pathways (mediated by phosphatidylinositol 3-kinase or PI 3-kinase, Akt, mTOR) and mitogenic pathways (mediated by Ras, Raf and mitogen-activated protein or MAP-kinase) pathways.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


