Wilson’s disease is found worldwide. The prevalence is approximately 1 in 30 000, with a carrier frequency of approximately 1 in 90 [3].
Molecular Genetics: Pathogenesis
The gene abnormal in Wilson’s disease, ATP7B, is on the long arm of chromosome 13. Its gene product is a copper-transporting P-type ATPase, the ‘Wilson ATPase’, which has six copper-binding units [4]. Both the gene and the Wilson ATPase have been characterized extensively [5,6]. The ATP7B gene is expressed mainly in the liver.
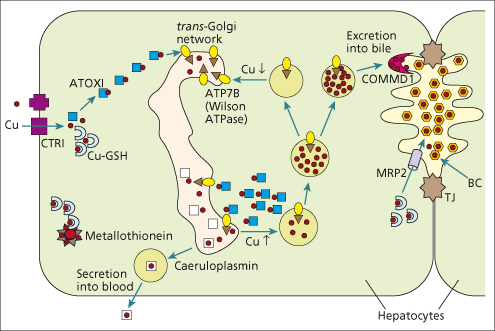
In normal hepatocytes, the Wilson ATPase is predominantly located in the trans-Golgi network into which it transports copper for incorporation into caeruloplasmin (Fig. 27.2). When intracellular copper concentrations are increased, the Wilson ATPase redistributes to a vesicular compartment close to the bile canalicular membrane [7,8]. Mutations in the Wilson ATPase may interfere with some or all of its complex functions in hepatocytes.
Fig. 27.2. Role of Wilson ATPase (ATP7B) in the hepatocellular disposition of copper. A doublet of hepatocytes is shown, with, on the right, the specialized bile canaliculus (BC) membrane located between two tight junctions (TJ). Starting at the left side of the diagram, copper (small red dots) is taken up across the sinusoidal plasma membrane by CTR1 (purple square), picked up and carried by ATOX1 (blue square) to the Wilson ATPase (yellow oval with arrow) in the trans-Golgi network. The Wilson ATPase either directs copper to production of caeruloplasmin or to excretion into bile, by a process which may also involve COMMD1. When intracellular copper concentrations are low or normal, the Wilson ATPase participates in production of holocaeruloplasmin (white square containing copper) in the Golgi apparatus; holocaeruloplasmin is then secreted into the blood. When intracellular copper concentrations are elevated, the Wilson ATPase expedites biliary excretion of copper. Glutathione (GSH; blue half circle) mediates other intracellular transfer, including incorporation into metallothionein and biliary excretion via MRP2. (Modified from Roberts EA, Sarkar B. Am. J. Clin. Nutr. 2008; 88: 851S–854S.)
Approximately 500 different mutations of ATP7B in Wilson’s disease have been identified (http://www.wilsondisease.med.ualberta.ca/database.asp). Most are point or missense mutations in the transmembrane and ATPase regions. Ideally, alterations in ATP7B must be shown to cause protein dysfunction to be counted as disease-causing mutations. These numerous mutations make mutation analysis impractical as a routine diagnostic test, except in areas where one or two mutations predominate, such as Eastern Europe, Sardinia, Iceland and the Canary Islands.
Phenotype–genotype correlations are difficult because most patients are compound heterozygotes, with a different mutation on each chromosome. No clear relationship has been found between the individual mutations and clinical disease patterns. Mutations that interfere with expression of the Wilson ATPase are associated with early liver disease. Homozygosity for the most common mutation in European populations (His1069Gln) may be associated with onset in adulthood and neurological form [9]. Phenotypic variation within families carrying the same mutation may be due to genetic modifiers of ATP7B, variations in other proteins involved in the hepatocellular handling of copper, compensatory cytoprotective mechanisms or environmental factors affecting copper availability. Onset of symptoms is significantly delayed in patients with the ApoEε 3/3 polymorphism [10].
Several animal models for Wilson’s disease exist. In mice, spontaneous point mutations in the murine Atp7b gene cause liver disease resembling human Wilson’s disease [11]; an Atp7b knockout mouse has more severe liver disease [12]. The Long-Evans Cinnamon (LEC) rat has a large deletion in the rat ATP7B. Homozygous rats show marked hepatic copper accumulation in the first few months of life with low serum caeruloplasmin, and they develop severe acute hepatitis. Surviving rats progress to chronic hepatitis and hepatocellular carcinoma [13]. D-penicillamine protects against these changes [14].
Because it is redox-active, copper can cause oxidative injury in cells [15]. Toxic levels of copper in the liver in Wilson’s disease cause alterations in hepatocellular mitochondria [16] and nuclei. Oxidative damage to mitochondria can be limited experimentally by vitamin E administration [17].
Pathology
Liver
The liver shows a spectrum of change from simple steatosis to periportal fibrosis to macronodular cirrhosis. With a fulminant hepatitis, submassive necrosis occurs.
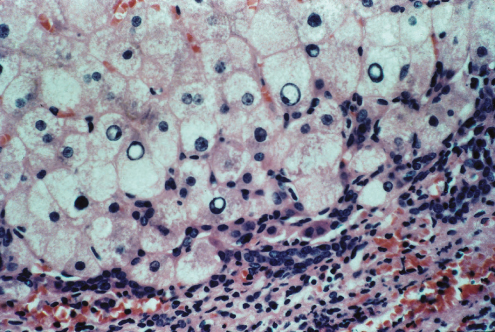
Liver cells are ballooned, show multiple nuclei, clumped glycogen and glycogenated nuclei (Fig. 27.3). Fatty change is usual. In some patients a particularly florid picture is seen with Mallory–Denk bodies, simulating acute alcoholic hepatitis. Alternatively, changes indicate chronic hepatitis (Fig. 27.4). Hepatic histology is not diagnostic, but in a young person with cirrhosis such a picture should always suggest Wilson’s disease.
Fig. 27.3. Hepatolenticular degeneration (Wilson’s disease). Liver cells adjoining a fibrous tissue band show gross vacuolation of their nuclei (glycogenic degeneration) and fatty change. (H & E, × 65.)
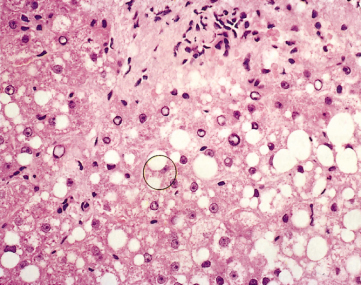
Fig. 27.4. Wilson’s disease. In this example there is piecemeal necrosis and lymphocytic infiltration as in autoimmune hepatitis. Note the hepatocellular swelling due to finely divided fat, and vacuolization of nuclei. (H & E, × 350.)
Rubeanic acid or rhodanine stains for copper are unreliable. In early disease, when copper is mainly in the cytoplasm, these stains do not detect any copper. Failure to stain copper on liver biopsy does not exclude the diagnosis of Wilson’s disease.
In cirrhosis, some nodules are positive whereas others are not. Detected copper is usually periportal, associated with atypical lipofuscin deposits. It must be distinguished from copper accumulation due to cholestasis.
Electron Microscopy
Mitochondria are large and pleiomorphic. They may contain ‘dark bodies’. The most striking change is cystic dilatation of the tips of the mitochondrial cristae. Mitochondria may be abnormal even in asymptomatic patients.
Other Organs
The Kayser–Fleischer ring is due to a copper-containing pigment deposited in Desçemet’s membrane at the periphery of the posterior surface of the cornea.
The kidney shows fatty and hydropic change with copper deposition in the proximal convoluted tubules.
Clinical Picture
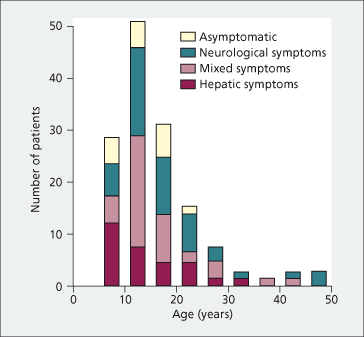
Clinical presentation is highly variable. It differs with age (Fig. 27.5). Most children present with liver disease (hepatic form). Later on, neuropsychiatric changes become increasingly predominant (neurological form). Patients presenting after age 20 usually have neurological symptoms [18,19]. The two forms frequently overlap. Most patients have developed symptoms or have been diagnosed between 5 and 40 years old. Numerous reports, however, indicate that some patients are diagnosed in early childhood or in their 50s–60s [20,21].
Fig. 27.5. Type of symptom complex at onset by age in 142 British and Chinese patients with Wilson’s disease [18].
The Kayser–Fleischer ring (Fig. 27.6) is a greenish-brown corneal ring appearing near the periphery of the iris. The upper pole is first affected. Expert slit lamp examination is almost always necessary to show it. It is usually present with neurological abnormalities but may be absent in 40–60% of those with a hepatic form. A similar ring may rarely be found with prolonged cholestasis and cryptogenic cirrhosis [22].
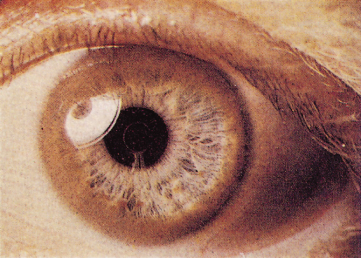
Rarely, the posterior layer of the capsule of the lens may show greyish-brown ‘sunflower’ cataracts, which resemble those due to copper-containing foreign bodies.
Hepatic Forms
Acute Liver Failure.
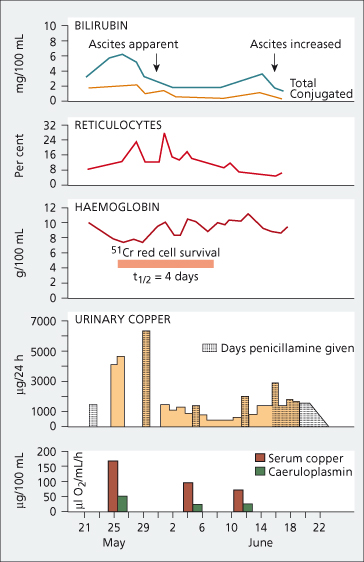
This is characterized by progressive jaundice, ascites and hepatic and renal failure, usually in a child or young person. The liver cell necrosis is presumably related to accumulation of copper. Virtually all patients are already cirrhotic. Acute intravascular haemolysis is due to destruction of erythrocytes by a sudden flux of copper from the necrotic hepatocytes (Fig. 27.7) [23]. Haemolysis of similar type is reported in sheep with copper intoxication, and in humans in accidental copper poisoning.
Fig. 27.7. Haemolytic crisis in Wilson’s disease, marked by a rise in serum (mainly unconjugated) bilirubin and followed by reticulocytosis. The haemoglobin fell and red cell survival was reduced. Urinary copper was very high even without the administration of penicillamine. Serum copper was higher than that usually found in Wilson’s disease. Ascites developed. The second episode of haemolysis, which was noted in June, was marked by a slight rise in serum bilirubin and a fall in haemoglobin [23].
Kayser–Fleischer rings may be absent. Urinary and serum copper levels are very high. Serum caeruloplasmin is usually low. However, it may be normal or raised since caeruloplasmin is an acute phase reactant, increased by active liver disease. Serum aminotransferases are inappropriately low for fulminant viral hepatitis and alkaline phosphatase may be very low [24]. In conventional American units, a low alkaline phosphatase to total bilirubin ratio (AP : TB less than 4) is highly suggestive of Wilson’s disease, more so when associated with aspartate aminotransferase : alanine aminotransferase (AST : ALT) more than 2.2 [25].
Autoimmune Hepatitis Mimic.
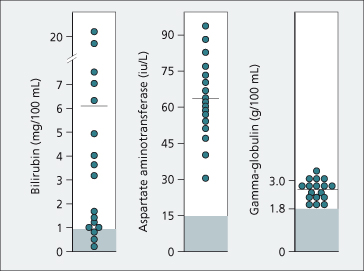
The condition presents at 10–30 years of age as an acute or chronic hepatitis with jaundice, elevated aminotransferases, hypergammaglobulinaemia and detectable non-specific autoantibodies (Fig. 27.8) [2,26]. Very rarely, Wilson’s disease and autoimmune hepatitis are coincidental. All such patients require investigation for Wilson’s disease.
Fig. 27.8. Biochemical tests in 17 patients with Wilson’s disease presenting as with chronic hepatitis resembling autoimmune hepatitis. Horizontal lines indicate mean values. Normal ranges are denoted by hatching (serum bilirubin 0.2–0.8 mg/dL; aspartate aminotransferase 4–15 IU/L; γ-globulin 0.7–1.8 g/dL) [2].
Cirrhosis.
The patient may present with insidiously developing cirrhosis, typically well-compensated. Clinical features include vascular spiders, splenomegaly, ascites and portal hypertension. Severe disease can exist without any neurological signs. Those showing evidence of hepatic decompensation may still respond well to medical treatment. Liver biopsy with measurement of hepatic copper concentration may be useful for diagnosis.
All children and younger adult patients with chronic liver disease should be screened for Wilson’s disease. Those showing any deterioration in school or work performance, slurring of the speech, change in handwriting, early ascites or transient haemolysis require extensive investigation. Any with a family history of cirrhosis should also be screened.
Hepatocellular carcinoma is uncommon [27]. Induction of hepatic metallothionein by copper may be protective [28].
Neuropsychiatric Forms
These broadly form subgroups according to the predominant features, and in order of incidence are: parkinsonian, pseudosclerotic, dystonic (dyskinetic) and choreic [29,30]. The neurological presentation may be acute and rapidly progressive. Early changes include a flexion–extension tremor of the wrists, grimacing, difficulty in writing, slurred speech and drooling. The limbs show a fluctuating rigidity. The intellect is fairly well preserved although patients with psychiatric disturbance usually present with a slow deterioration of the personality or depression. Approximately 15% of all patients present with psychiatric disturbance only.
Often the neurological changes are chronic. Onset is in early adult life with tremor, marked and of a wing-beating type, exaggerated by voluntary movement. Sensory loss and pyramidal tract signs are absent. The expression is vacant. Severely dystonic patients have a worse prognosis than other groups [31]. Among patients with neurological forms, 20% may only have minimal changes or steatosis on liver biopsy.
The electroencephalogram shows generalized non-specific changes which may also be seen in asymptomatic siblings. Presentation as a seizure disorder is rare.
Renal Changes
Aminoaciduria, glycosuria, phosphaturia and uricosuria reflect renal tubular changes. These are presumably due to copper deposition in the proximal renal tubules.
Renal tubular acidosis is frequent and may be related to stone formation.
Other Changes
Isolated episodes of haemolysis may occur; these resolve spontaneously. Rarely, the lunulae of the nails appear lavender due to increased copper. Skeletal changes include demineralization, premature osteoarthritis, subarticular cysts and fragmentation of bone about the joints. Presenting symptoms may occasionally be limited to arthralgias and muscle weakness. Gallstones are related to haemolysis. Hypoparathyroidism is a rare association. Cardiac disease, in particular dysrhythmias, has been reported. Decreased fertility in women or repeated spontaneous abortions may occur.
Laboratory Tests
Serum caeruloplasmin and copper levels are usually low [18,32–34]. Other causes of low caeruloplasmin include the genetic disorder acaeruloplasminaemia [35], advanced liver disease with synthetic failure and severe malnutrition. The caeruloplasmin level may be raised by inflammation, oestrogen administration, oral contraceptive drugs, biliary obstruction or pregnancy.
Basal 24-h urinary copper excretion is increased (>0.6 µmol/24 h). This highly informative test requires some attention to methodology. Wide-necked bottles with copper-free disposable polyethylene liners have been recommended. Simultaneous measurement of urinary creatinine validates completeness of collection. Up to three separate collections should be performed.
Incorporation of orally administered radio-copper into caeruloplasmin is no longer used for diagnosis.
Liver Biopsy
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


