Hepcidin, expressed in many cell types involved in iron transport, binds to ferroportin. The precise mechanism by which HFE protein, ferroportin and hepcidin interact is unclear. Normally, the response to excess body iron would be increased hepatic expression of hepcidin. The observed deficiency (absolute or relative to that expected) of hepcidin is a unifying explanation for increased iron absorption and iron overload observed in many hereditary forms of haemochromatosis, especially those associated with missense mutations in genes that encode HFE (HFE), transferrin receptor-2 (TFR2), hepcidin (HAMP) and haemojuvelin (HJV) [7,8].
Circulating Iron
In the plasma, iron is bound to transferrin, a glycoprotein largely synthesized in the liver. Transferrin can bind two ferric iron molecules, and is responsible for the ‘total iron-binding capacity’ of serum of 250–370 µg/dL. This is normally around 20–40% saturated with iron. Physiological entry of iron into reticulocytes and hepatocytes depends upon transferrin receptors at the cell surface, which preferentially bind transferrin carrying iron. The receptor/iron transferrin complex is internalized and the iron released. This process is saturable. TfRs are down-regulated as the cell becomes replete with iron.
When serum transferrin is fully saturated, as in overt haemochromatosis, iron circulates also in ‘non-transferrin bound’ forms, associated with low-molecular-weight chelators. Iron in this form readily enters cells by a non-saturable process.
Storage of Iron
Iron is stored in cells in ferritin, a combination of the protein apoferritin (H and L subunits) and iron. High concentrations of iron stimulate apoferritin synthesis. Up to 4500 atoms of iron can be stored within a single ferritin molecule. Aggregates of degraded ferritin molecules make up haemosiderin, which stains as blue granules with ferrocyanide. Lipofuscin, a yellow brown material may accumulate in association with iron overload, but does not contain iron.
Iron contained in cells as ferritin or haemosiderin is available for mobilization. The normal total body content of iron is about 4 g, of which 3 g is present in haemoglobin, myoglobin, catalase and other respiratory enzymes. Storage iron comprises 0.5 g; of this 0.3 g is in the liver but is not seen with the usual histological stains for iron. The liver is the predominant site for storage of iron absorbed from the intestine. When its capacity is exceeded, iron is deposited in other parenchymal cells, including the acinar cells of the pancreas, and the cells of the anterior pituitary gland. The reticuloendothelial system plays only a limited part in iron storage unless this is the result of transfusion, when it is concentrated particularly in the spleen.
Iron Overload and Liver Damage
Fibrosis and hepatocellular damage are directly related to the iron content of the liver cell. The pattern of damage is similar irrespective of whether the overload is due to genetic haemochromatosis or to multiple transfusions. The severity of fibrosis is maximal in periportal areas where iron is particularly deposited.
When iron deposition is low it is stored as ferritin. As the load increases more is present as haemosiderin.
Removal of iron by venesection or chelation leads to clinical and biochemical improvement with reduction or prevention of hepatic fibrosis [9,10].
There are several processes by which iron can damage the liver. There is enhanced oxidative stress in patients with iron overload and this is associated with increased TGF-β1 expression. Oxidative stress causes lipid peroxidation of membranes of organelles leading to functional defects of lysosomes, mitochondria and microsomes. Mitochondrial cytochrome C oxidase activity is reduced. There is lysosomal membrane fragility and release of hydrolytic enzymes into the cytosol.
Hepatic stellate cells (lipocytes) are activated in genetic haemochromatosis and activation is reversed by iron removal. Stellate cell activation appears related to the release of cytokines and other substances from neighbouring cells rather than oxidant stress within stellate cells [11].
Genetic Haemochromatosis
In 1865, Trousseau described the clinical syndrome of skin pigmentation, cirrhosis and diabetes now recognized as characteristic of late-stage genetic haemochromatosis. This is an autosomal recessive metabolic disorder in which there may be increased iron absorption over many years.
Molecular Genetics
Sheldon in his classic monograph described idiopathic haemochromatosis as an inborn error of metabolism [12]. The discovery of genetic linkage of haemochromatosis to the HLA serotype allowed the inheritance to be defined as autosomal recessive, and placed the gene on chromosome 6.
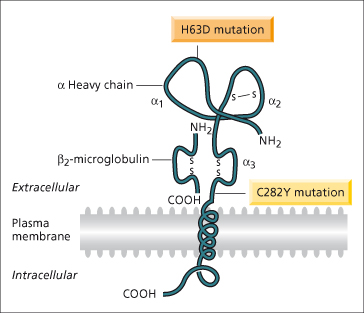
In 1996, a positional cloning approach was successful in identifying the HFE gene approximately 6 megabases telomeric to the HLA-A locus on chromosome 6 [1] (Fig. 26.2). Eighty-five per cent of chromosomes from haemochromatosis patients contained a single mutation (C282Y, also designated Cys282Tyr) in the HFE gene compared with 3% of control chromosomes. In most populations of northern European origin, over 90% of haemochromatosis patients have been found to be homozygous for this mutation [13]. In southern European populations, the frequency of C282Y homozygosity is lower (65%) [14]. The high frequency of this mutation in genetic haemochromatosis points to individuals being descended from a single family or community (probably Celtic) in which the mutation initially occurred [15]. A second mutation described at the time of the discovery of the HFE gene (H63D; also known as His63Asp) is common in the normal population.
Fig. 26.2. Hypothetical model of the HFE protein based on homology with MHC molecules. The extracellular component has three α-domains, one of which binds to β2-microglobulin, a membrane spanning region and a short cytoplasmic tail. The C282Y mutation disrupts the disulphide bond in the α3-domain through the substitution of tyrosine for cysteine. The H63D mutation is in the α1-domains.
(Modified from Feder JN et al. [1] with permission.)
The frequency of C282Y homozygosity found in population screening studies is 1 in 200–300 [16–18]. This frequency, however, does not correspond to the frequency of clinically recognized haemochromatosis. Although biochemical penetrance (raised ferritin, transferrin saturation) is found in 50–80% of susceptible individuals, disease penetrance (i.e. symptoms, hepatic fibrosis/ cirrhosis) is low [19]. It has been demonstrated that approximately 28% of male C282Y homozygotes, but only 1% of female C282Y homozygotes, may have a haemochromatosis-related symptom [19].
The contribution of the H63D mutation to iron overload is unclear and the effect, if any, appears to have a low penetrance. Focus has mainly been on compound heterozygotes (C282Y/H63D) and H63D homozygotes, where it has been estimated that approximately 1.5% will develop significant iron overload [16].
Heterozygotes
The frequency of heterozygosity for the C282Y mutation in populations of Northern European origin is approximately 10%. Although heterozygotes have mean serum iron and transferrin saturation values higher than normal subjects, significant iron overload is extremely rare. However, since these individuals may have slight increases in intracellular iron it has been questioned whether this would enhance damage from other diseases. Hepatic fibrosis/cirrhosis due to hepatitis C or alcohol, however, has not been found to be worsened by heterozygosity for C282Y [20].
Pathology
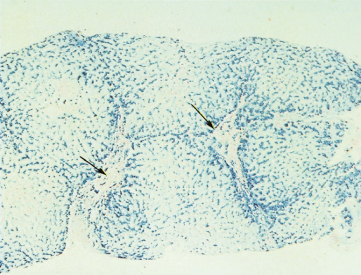
The liver in the early stages may show only portal zone fibrosis with deposition of iron in the periportal liver cells and, to a lesser extent, in the Kupffer cells. Fibrous septa then surround groups of lobules and irregularly shaped nodules (holly leaf appearance). There is partial preservation of the architecture, although ultimately a macronodular cirrhosis develops (Fig. 26.3). Fatty change is unusual and the glycogen content of the liver cells is normal.
Fig. 26.3. The liver in genetic haemochromatosis. Cirrhosis is seen and hepatocytes are filled with blue-staining iron pigment. Fibrous tissue is also infiltrated with iron. The arrows indicate portal tracts. (Perls’ strain, × 13.)
Cirrhotic patients with iron-free foci have a higher risk of developing hepatocellular carcinoma [21].
Iron deposition in organs beyond the liver is only seen with severe iron overload.
The pancreas may show fibrosis and parenchymal degeneration with iron deposition in acinar cells, macrophages, islets of Langerhans and fibrous tissue.
Heart muscle may be involved, muscle fibres being replaced by a mass of iron pigment within the sheath. Degeneration of the fibres is rare.
Spleen, bone marrow and duodenal epithelium do not show the iron overload seen elsewhere. Brain and nervous tissue are also usually free of iron.
Epidermal atrophy may reduce the skin to a flattened sheet. Hair follicles and sebaceous glands are inconspicuous. Characteristically, the melanin content of the basal layer is increased. Iron is usually absent from the epidermis but can often be seen deeper, especially in the basal layer.
Endocrine glands, including adrenal cortex, anterior lobe of pituitary and thyroid, may show varying amounts of iron and fibrosis.
The testes are small and soft with atrophy of the germinal epithelium without iron overload. There may be interstitial fibrosis and iron is found in the walls of capillaries.
Relation to Alcoholism
In an experimental model of alcoholic liver disease, the addition of iron to the diet results in cirrhosis. In patients, the combination of haemochromatosis and excess alcohol intake results in more advanced liver disease [22]. Hepatic iron deposition is recognized in alcoholic liver disease (as well as other end-stage liver disease) and may be due to increased intestinal iron absorption in chronic alcohol abusers. Ethanol decreases hepcidin expression in both in vitro cellular studies and in vivo models [23], and this would be expected to increase iron absorption.
Clinical Features
The classical picture is of a lethargic, middle-aged man with pigmentation, hepatomegaly, diminished sexual activity, loss of body hair and arthralgia; diabetes is common. This picture is seen in a minority of C282Y homozygotes. An asymptomatic patient is the most common scenario [24].
Diagnosis depends on a high degree of suspicion and should be considered in any patient with symptomless hepatomegaly and virtually normal biochemical tests of liver function [25]. In view of the C282Y homozygote frequency found in the community, the condition must be considerably more frequent than is recognized. There is a mean delay of 5–8 years between presentation and diagnosis [24].
Overt haemochromatosis is 10 times more frequent in males than females [26]. Women are spared by iron loss with menstruation and pregnancy. Female patients with haemochromatosis usually, but not always, have absent or scanty menstruation, have had a hysterectomy or are many years postmenopausal.
Haemochromatosis is rarely diagnosed before the age of 20, and the peak incidence is between 40 and 60 years.
The slate grey pigmentation when present is maximal in the axillae, groins, genitalia, old scars and exposed parts. It can occur in the mouth. The colour, due to increased melanin in the basal layer, appears through the atrophied, superficial epidermis. The skin is shiny, thin and dry.
Hepatic Changes
The liver may be enlarged and firm. Abdominal pain, usually a dull ache with hepatic tenderness, is noted in 56% of cases [26].
Signs of hepatocellular failure are usually absent and ascites rare. The spleen is palpable but rarely large. Bleeding from oesophageal varices is unusual.
Primary liver cancer develops in 15–30% of cirrhotic patients [27–29]. It may be the mode of presentation, particularly in the elderly. It should be suspected if the patient shows clinical deterioration with rapid liver enlargement, abdominal pain and ascites. Serum α-fetoprotein may be increased.
Endocrine Changes
At diagnosis, about 70% of cirrhotic patients, but only 17% of non-cirrhotic patients, have clinical diabetes [29]. This may be complicated by nephropathy, neuropathy, peripheral vascular disease and proliferative retinopathy. The diabetes may be easy to control or may be resistant to large doses of insulin. It could be related to a family history of diabetes, to cirrhosis of the liver which impairs glucose tolerance or to direct damage to the pancreas by iron deposition. In population screening studies of asymptomatic C282Y homozygotes, the prevalence of diabetes did not differ from the control population [16, 30].
Loss of libido or potency occurs in approximately 35% of patients and amenorrhoea in 15% of women [29]. Hypogonadism may be due to hypothalamic, pituitary or gonadal dysfunction or a combination of all three [31].
Pituitary function is impaired to a variable extent in about two-thirds of patients. This is related to iron deposition in the anterior pituitary. Gonadotrophin-producing cells are selectively affected. Hypogonadotrophic testicular failure is shown by impotence, loss of libido, testicular atrophy, skin atrophy and loss of secondary sexual hair. Plasma testosterone levels are subnormal. Testosterone levels increase following administration of gonadotrophins, suggesting that the testes are capable of responding.
Osteoporosis is seen particularly when hypogonadism is present [32].
Panhypopituitarism with hypothyroidism and adrenal corticodeficiency are rarer.
Cardiac Changes
Changes on ECG are reported in 35% of patients presenting with haemochromatosis [29]. Echocardiographic abnormalities are also seen, are related to the degree of iron overload and improve with venesection [33]. Presentation with heart failure, particularly in younger subjects, is seen but is unusual. The picture is of progressive right-sided heart failure, sometimes with sudden death. The ‘iron heart’ is a weak one. Dysrhythmias are also seen.
Cardiac complications are presumably related to iron deposits in the myocardium and conducting system.
Arthropathy
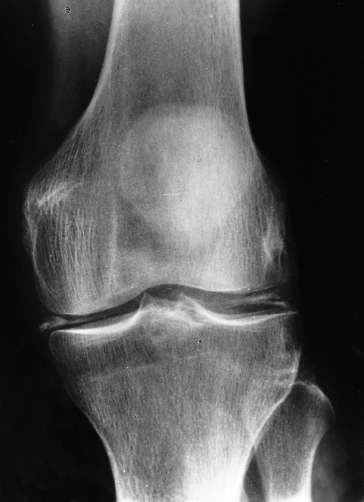
In about two-thirds of patients, a specific arthropathy starts in the metacarpophalangeal joints (Fig. 26.4) [34]. Wrists and hips may also be affected [35]. It may be a presenting feature. Radiologically there is a hypertrophic osteoarthritis. Chondrocalcinosis is seen in the menisci and articular cartilage (Fig. 26.5). It is related to an acute crystal synovitis with calcium pyrophosphate.
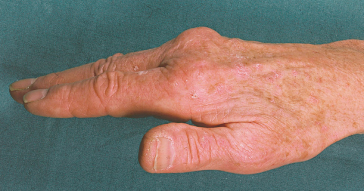
Fig. 26.5. Genetic haemochromatosis. Radiograph of the knee joint shows chondrocalcinosis in menisci and articular cartilage.
(Courtesy of M. Barry.)
Arthralgia is often the most difficult long-term clinical problem as it is resistant to conventional anti-inflammatory agents. It is present in 45% of patients at diagnosis. After depletion of body iron, 30% improve but in 20% symptoms worsen [29].
Special Investigations
Transferrin Saturation
In HFE-related haemochromatosis the serum iron is increased and the transferrin concentration/total iron binding capacity reduced, giving a high transferrin saturation. In those with an increased serum ferritin, this may be up to 100%. A normal transferrin saturation in the presence of a high serum ferritin should lead to consideration of other causes of hyperferritinaemia such as inflammation, metabolic syndrome with hepatic steatosis, alcohol excess and, rarely, non-HFE inherited iron overload, in particular ferroportin disease.
Serum Ferritin
Ferritin is the major cellular iron storage protein. The form present in normal serum contains little iron. Its function there is uncertain. The serum concentration is proportional to body iron stores (Fig. 26.6). It is of value in assessing body iron stores [36–38], but can be unreliable in early diagnosis at the precirrhotic stage, and in patients with hepatic inflammation and a raised transaminase. It is useful in following treatment.
Fig. 26.6. Natural history of untreated genetic haemochromatosis. Relationship between the serum ferritin and the progression of events leading to the clinical syndrome [38].
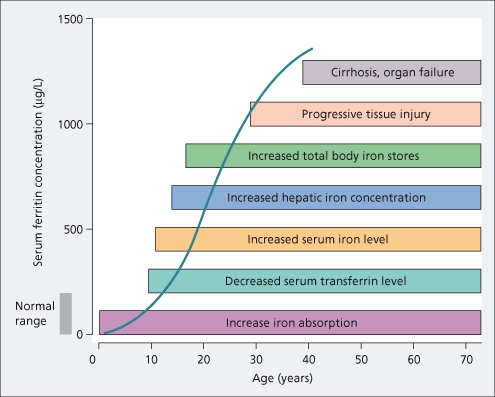
With hepatocellular necrosis, serum ferritin increases as it is released from liver cells [38]. High serum ferritin levels are also seen with inflammatory conditions, such as hepatitis, alcohol excess, fatty liver and some cancers.
Needle Liver Biopsy
Since the introduction of mutation analysis for the HFE gene, the indication for needle liver biopsy has changed. Previously, hepatic histology and iron quantification was important for diagnosis, giving an indication of the severity and pattern of iron deposition. Measurement of liver iron was important for calculation of the liver iron index (the liver iron concentration divided by the age of the patient), which was of diagnostic value in genetic haemochromatosis. Since mutation analysis confirms the diagnosis in the majority of cases, liver biopsy is only necessary in C282Y homozygotes to assess whether there is severe fibrosis or cirrhosis (see Fig. 26.3), which determines the protocol for subsequent follow-up. Analysis of risk factors has shown that cirrhosis is unlikely in patients without hepatomegaly, with a normal alanine transaminase and a serum ferritin of less than 1000 µg/L [39]. The current recommendation is that in the absence of these features, liver biopsy is not necessary. If any of these features are present then liver biopsy is recommended since there is an approximate 50% chance of severe fibrosis or cirrhosis [40]. Hepatic elastography may be a useful tool for the non-invasive assessment of cirrhosis [41].
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


