The most distinctive aspect of the biology of this virus is its replication. Unlike all other RNA viruses, HDV lacks its own RNA polymerase. Thus, for replication HDV exploits a host cellular enzyme, the host RNA polymerase (Pol) II, which normally copies double-stranded DNA templates. HDV has the unique ability to redirect this cellular enzyme to transcribe the HDV RNA genome [8]. The only enzymatic activity that is inherent to HDV is mediated by RNA elements termed ribozymes (RNA enzymes), which cleave the circular RNA genome producing a linear molecule [3]. The HDV ribozyme is the only catalytic RNA motif discovered in humans.
HDV Genotypes
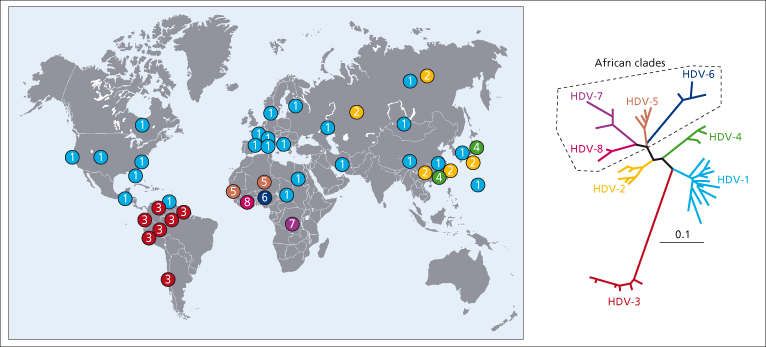
Like all RNA viruses, HDV is characterized by a high degree of genetic heterogeneity. Nucleotide sequences of HDV isolates obtained worldwide indicated the existence of eight genotypes, varying by as much as 38% over their entire genomic sequences [9,10]. The HDV genotypes differ in their geographic distribution (Fig. 19.2). While genotype 1 is detected worldwide, genotypes 2, 3 and 4 are more geographically restricted. Genotypes 2 and 4 (previously termed IIa and IIb) are found primarily in the Far East; genotype 3 exclusively in the northern regions of South America. More recently, four new HDV genotypes have been detected in West and Central Africa (5, 6, 7 and 8) [9,10]. Evidence for a mixed HDV infection and HDV genome recombination has been recently described, providing a novel mechanism for HDV evolution [10].
Epidemiology
Transmission
HDV can be transmitted only to individuals who simultaneously harbour HBV. Persons with hepatitis B surface antibody (anti-HBs), being immune to HBV infection, are not susceptible to HDV. Transmission of HDV and HBV may occur simultaneously (coinfection) or HDV may infect a chronic HBsAg carrier (superinfection). The routes of HDV transmission are the same as for HBV, the most important being parenteral exposure to blood or blood products, either overtly or through the inapparent parenteral route involving interpersonal contacts. HDV can affect health-care workers, transfusion recipients and haemophiliacs. Intrafamily spread has been documented, both in adults and in children. Sexual transmission may occur, particularly among sexually promiscuous groups, but HDV infection is infrequent in homosexual men. Vertical transmission is rare.
Geographic Distribution
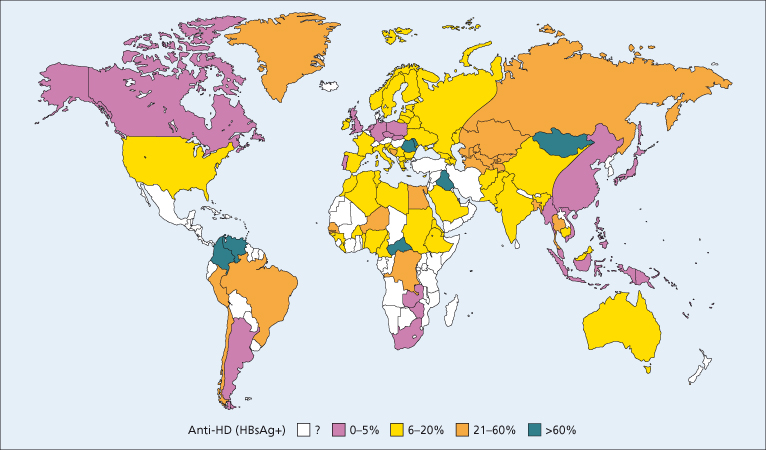
Infection with HDV has a global distribution with considerable geographic variation (Fig. 19.3). It has been estimated that 15 million HBsAg carriers are infected with HDV worldwide [11]. Seroepidemiological studies performed in the 1980s showed that the prevalence of HDV infection is high in the Mediterranean area, the Middle East, Central and Northern Asia, East Africa, the Amazon Basin and certain areas of the Pacific, whereas it is low in North America and Northern Europe, South Africa and Eastern Asia (Fig. 19.3) [3]. The high rate in Southern Europe is the result of a mixed epidemiological pattern characterized by endemic infection in the general population and epidemic outbreaks among intravenous drug users, whereas in industrialized countries HDV infection is mainly confined to intravenous drug users, although it can affect all groups that are at risk for HBV infection [3]. In developed countries, universal screening for HBsAg of blood and its derivatives has led to the virtual elimination of hepatitis D transmission through blood transfusion.
Fig. 19.3. Worldwide prevalence of HDV infection as measured by anti-HD in HBsAg carriers with acute or chronic hepatitis.
(Adapted from [3] with permission.)
Epidemics of HDV infection, including outbreaks of fulminant hepatitis, have been reported in the Amazon Basin, Brazil (Labrea fever), Ecuador, Colombia (Santa Marta hepatitis), Venezuela and Equatorial Africa [12]. In these areas, children of the indigent population are affected and the mortality is high.
Changing Epidemiology
Over the past two decades, there has been a significant decline in the prevalence of HDV infection along with HBV, particularly In Italy where the HDV prevalence in HBV carriers with liver disease has declined from 25% in 1987 to 14% in 1992 and 8% in 1997 [13]. Likewise, the incidence of acute hepatitis D in Italy dropped from 3.1 to 0.2 per 106 inhabitants per year [11]. This is probably the result of three events: the universal HBV vaccination programmes, the improvement in hygiene and the AIDS campaigns, which addresses the dangers of promiscuity and of shared syringes and needles. However, in parallel with a decline in areas that were previously endemic, new foci of HDV infection have emerged in other regions of the world, such as South-eastern Russia, Okinawa, Northern India and Albania (11). Moreover, immigration is posing a new threat for HDV resurgence in Europe. Recent epidemiological studies conducted in Germany [14], France [15], Italy [16] and England [17] have shown that the prevalence of HDV infection from the late 1990s has not further decreased in these countries. Overall, these epidemiological studies suggest that the reservoir of HDV in Europe is sustained by two different pools of HDV-infected individuals [13]. One is composed of individuals who survived the HDV epidemic in the 1970s and 1980s and the other is represented by young subjects who migrated to Europe from areas where HDV infection is endemic.
Pathogenesis
The liver is the only organ in which HDV can replicate. The virus is not directly cytopathic, and there is evidence that the liver damage is immunologically mediated [3]. The peak replication of HDV precedes the peak of the histopathological changes.
The mechanisms whereby HDV causes the most rapidly progressive form of liver fibrosis are unknown. Recently, it has been suggested that the long form of HDAg may play a role in HDV pathogenesis by inducing liver fibrosis through the regulation of transforming growth factor-β-induced signal activation [18]. This regulation is accomplished by isoprenylation of the long HDAg.
The role of the HDV genotypes in the pathogenesis of hepatitis D remains to be elucidated, although reported studies indicate that the genotype of HDV may influence the disease severity. Genotype 1, the most prevalent worldwide, has been associated with a broad spectrum of pathogenicity. In Taiwan, patients infected with genotype 1 had a lower remission rate and more adverse outcomes than those with genotype 2 [19]. Genotypes 2 and 4 have been associated with milder forms of acute and chronic hepatitis D [20], while genotype 3 has been associated with outbreaks of fulminant hepatitis in South America [21]. Genotype 3 is associated exclusively with the HBV genotype F, while a less restricted association has been documented between the other HDV genotypes and the HBV genotypes [10]. Further studies are needed to investigate whether biological differences exist among the HDV genotypes, the contribution of the coinfecting HBV genotypes and the variability of the host immune response.
Modes of Infection and Clinical Course
Acute Hepatitis D
Coinfection (Fig. 19.4)
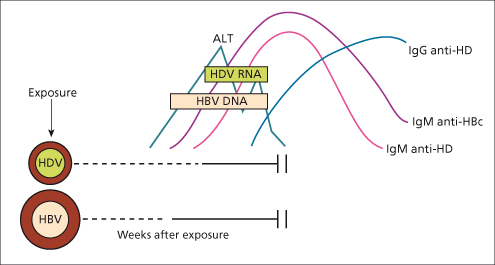
The clinical expression of acute hepatitis D acquired by coinfection with HBV may range from mild to severe or even fulminant hepatitis. In most cases, acute hepatitis D is self-limited as HDV cannot outlive the transient HBs antigenaemia. The clinical picture is usually indistinguishable from acute hepatitis B alone. However, a biphasic rise in alanine and aspartate aminotransferases occurring a few weeks apart may be noted, the second rise being due to the acute effects of HDV. The outcome is a complete recovery, as typically seen in hepatitis B, and in only 2% of cases it progresses to chronicity [22]. The long-term outcome is therefore good.
Fig. 19.4. Simultaneous infection with HBV and HDV results in acute hepatitis B with a rise in alanine transaminase (ALT) and the appearance of IgM anti-hepatitis B core. HDV infection follows with a second peak of ALT and the appearance of IgM anti-delta (anti-HD) in serum. Clearing of HBV is associated with clearing of HDV
[11].
Superinfection
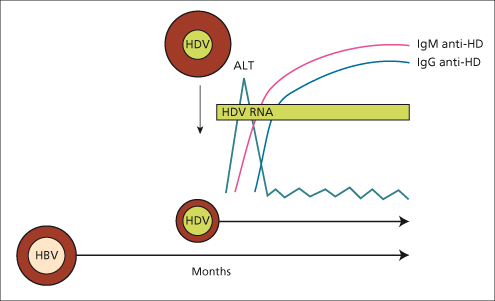
(Fig. 19.5)In the superinfection pattern, the pre-existing HBV infection provides the ideal background for the full expression of HDV. This results in an overt, severe acute hepatitis, which may run a fulminant course. The acute clinical attack is usually marked by jaundice. It may present as an exacerbation of a pre-existing chronic hepatitis B, leading rapidly to liver failure, or as a new hepatitis in a previously asymptomatic HBsAg carrier. If the HBsAg state is unknown it may be misdiagnosed as acute hepatitis B [23]. Therefore, HDV infection should be considered in any HBsAg-positive subject with acute or fulminant hepatitis, the latter being more common in hepatitis D than in the other types of viral hepatitis [24]. Since the HBsAg-carrier state permits the continuous replication of HDV, superinfection of acute hepatitis D leads to chronic hepatitis D in over 90% of cases.
Fig. 19.5. HDV superinfection of a chronic HBsAg carrier results in an attack of acute hepatitis with the appearance of IgM anti-delta (anti-HD) followed by IgG anti-delta (anti-HD). The pre-existing HBV infection provides continuous support to HDV replication. This results in acute hepatitis progressing to chronicity in over 90% of the cases [11].
Chronic Hepatitis D
The clinical presentation of chronic hepatitis D varies considerably. It may be virtually symptom free and discovered incidentally after a routine medical check, or it may present with symptoms. The most common is fatigue, but malaise, anorexia, right upper quadrant discomfort and dark urine can occur, especially if the disease is severe or advanced. Some may present with the complications of established cirrhosis with jaundice, encephalopathy, ascites or portal hypertension. There are no specific clinical features, although, at variance with chronic hepatitis B, half the patients with chronic hepatitis D report a history of a previous attack of acute hepatitis, which represents the time of superinfection with HDV. Many exhibit splenomegaly. The disease is severe at all ages, including children [25,26]. Typically, patients with chronic hepatitis D have persistently high serum aminotransferase levels, which tend to decrease as the disease progresses to the late stage of cirrhosis. Most patients are hepatitis B e antibody (anti-HBe)-positive and have low or undetectable levels of serum HBV DNA, supporting the concept that the liver damage is caused by HDV. Chronic hepatitis D should always be suspected in HBsAg carriers with active liver disease and undetectable serum HBV DNA.
Laboratory test abnormalities in chronic hepatitis D are similar to those detected in chronic hepatitis B alone, with the exception in the former of higher levels of gammaglobulins. Patients with chronic hepatitis D may develop a variety of autoantibodies. About 5% of them show autoantibodies against the microsomal membrane of the liver and kidney (LKM-3) [11].
Hepatic Histology
There are no distinctive histological features that differentiate hepatitis D from the other forms of viral hepatitis, except that hepatitis D tends to be more severe. The morphological hepatic changes, consisting of hepatocellular necrosis and inflammation, are those typical of acute or chronic viral hepatitis. In acute hepatitis D, pathological changes are often focal with a prominent intralobular infiltration of inflammatory cells, mainly lymphocytes and macrophages, and a degenerative cytoplasmic eosinophilia leading to the formation of acidophilic bodies in the parenchyma and portal tracts [27]. In the most severe cases, including fulminant hepatitis, confluent necrosis may involve dropout of most but not all hepatocytes (submassive necrosis) or virtually all hepatocytes (massive necrosis). In chronic hepatitis D, the degree of periportal necrosis (interface hepatitis) is usually more prominent than that seen in other forms of chronic viral hepatitis (Fig. 19.6) and it is often accompanied by active micronodular and macronodular cirrhosis.
Fig. 19.6. Chronic hepatitis D showing marked periportal (interphase) and lobular inflammatory activity. Aggregates of Kuffper cells and lymphocytes are seen around hydropic and necrotic hepatocytes (H&E, × 200).
(Courtesy of Dr Sugantha Govindarajan.)
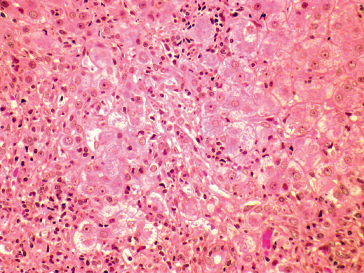
A peculiar histological feature observed in epidemics of fulminant hepatitis D in the Amazon Basin is microvesicular steatosis of the hepatocytes which leads to the formation of morula cells (Fig. 19.7) [28]. Similar alterations, however, have also been observed in severe hepatitis D elsewhere, in Italy and Africa [3,11].
Fig. 19.7. Fulminant hepatitis D (Labrea hepatitis) in a 3-year-old girl from Northern Brazil who died after 3 days of symptoms. An autopsy liver sample shows microvesicular fatty change in large hepatocytes with central nucleus (Morular, plant-like cells). (Immunoperoxidase, × 500.)
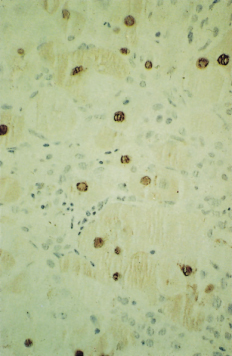
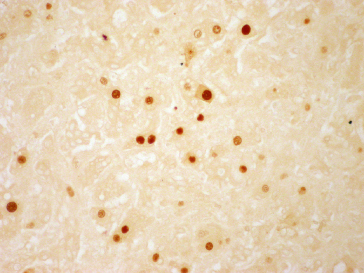
Detection of HDAg in the Liver
Using immunofluorescence or immunohistochemistry, HDAg may be shown in the nuclei of hepatocytes (Fig. 19.8). The presence of HDAg increases from acute to chronic hepatitis but decreases as the disease progresses to the late stage of cirrhosis. Thus, it may give false-negative results in patients with advanced disease.
Fig. 19.8. Immunoperoxidase staining showing hepatitis delta antigen in the hepatocyte nuclei (H&E, × 200).
(Courtesy of Dr Sugantha Govindarajan.)
Course and Prognosis
The course of chronic HDV infection varies considerably. Chronic hepatitis D is the least common but the most severe and rapidly progressive form of viral hepatitis at all ages. Cirrhosis develops in 70 to 80% of the cases within 5 to 10 years [29], and at a younger age, more than one decade earlier, than in HDV-negative HBV cirrhosis [30]. The risk of developing cirrhosis is about threefold higher in HDV-infected patients compared to those with HBV alone. Once established, HDV cirrhosis may be a stable disease for many years, but a high proportion of patients die of the complications of cirrhosis, end-stage liver disease and hepatocellular carcinoma (HCC), unless they receive liver transplantation. The proportion of patients who ultimately develop each of these complications is not well defined because of the difficulty in recruiting large cohorts of patients with chronic hepatitis D followed prospectively at regular intervals. Most of the data on complications rates have been inferred from retrospective studies; yet, they have provided an overall picture of the natural history of chronic hepatitis D. It has been estimated that the annual incidence of liver decompensation in HDV cirrhotic patients ranges from 2.6 to 3.6% and that of HCC from 2.6 to 2.8% [30,31]. In a recent longitudinal study conducted in Italy, liver decompensation was the most frequent cause of death in HDV cirrhosis [31]. In a longitudinal analysis of 200 Western European patients with compensated HBV cirrhosis, including 39 coinfected with HDV, the adjusted relative risk of HCC was threefold and that of decompensation and mortality twofold higher in HDV cirrhosis than in HBV cirrhosis during a median follow-up of 6.6 years. The cumulative 10-year survival free of liver transplantation is higher for patients with mild chronic hepatitis (100%) or severe chronic hepatitis (90%) than for those with histological (asymptomatic) cirrhosis (58%) or clinical cirrhosis (40%) [32]. As the disease progresses to end-stage liver disease, both alanine aminotransferase (ALT) levels and HDV replication tend to decrease. The mean interval between primary HDV infection and histological cirrhosis is about one decade, but once cirrhosis is established the disease is stable and compatible with a good quality of life for another decade. Thus, the mean interval between primary infection and liver decompensation is about two decades [32], although more rapidly progressive forms have been described [11].
In a minority of cases, HDV infection is marked by a non-progressive, benign course, as reported in some populations in endemic areas, such as the Greek community of Archangelos and the American Samoa [33]. Disease resolution may also occur spontaneously in the natural history of HDV infection, with a higher rate of spontaneous HBsAg clearance among HDV-positive than HDV-negative HBV carriers [34]. However, adverse clinical outcomes such as liver decompensation, HCC or death may occur if HBsAg clearance takes place after HDV disease has progressed to advanced stages.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


