Learning points
- Fibropolycystic liver diseases are heterogeneous, overlapping conditions in which liver cystic lesions are often associated with renal abnormalities.
- There is a wide spectrum of polycystic disease (differing in age of presentation, predominantly renal or liver pathology) based on whether inheritance is autosomal recessive or dominant.
- Congenital hepatic fibrosis (CHF) is mainly diagnosed in childhood or adolescence, with features of portal hypertension or with the incidental finding of isolated firm hepatomegaly. Often there is associated renal pathology.
- Caroli’s disease is characterized by congenital, segmental, saccular dilatations of the intrahepatic bile ducts without other hepatic abnormalities. When associated with CHF, it is called Caroli’s syndrome.
- Recurrent cholangitis is a serious complication of autosomal recessive polycystic kidney disease, Caroli’s disease and CHF, leading to deteriorating liver damage and liver failure.
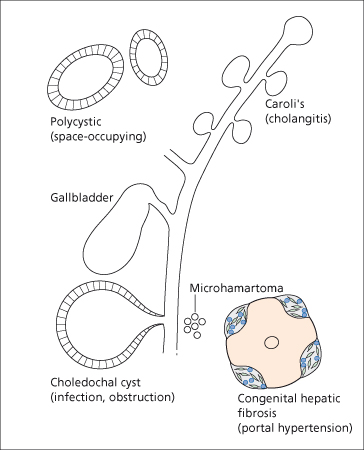
Cystic lesions of the liver and bile ducts are increasingly being diagnosed because of improved imaging techniques. They comprise several different conditions, some hereditary, some sporadic, some presenting mainly in childhood, others typically in adult life. Liver and bile duct cystic lesions may overlap (Fig. 14.1). According to the underlying pathology, clinical symptoms are variable and encompass those of recurrent jaundice, of a space-occupying lesion, of portal hypertension and of cholangitis (Fig. 14.2).
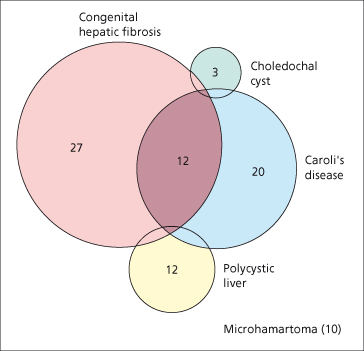
Fig. 14.1. Venn diagram showing one series of 51 patients in which many had more than one fibropolycystic disease. The combination of congenital hepatic fibrosis and Caroli’s disease was most striking. Microhamartomas, although reported in only 10 patients in this series, are common [53].
Fibropolycystic Diseases
Fibropolycystic diseases of the liver comprise a heterogeneous group of genetic disorders in which segmental dilatations of the intrahepatic bile ducts are associated with fibrosis. Although classified into specific conditions, there is much overlap with a varied contribution of microscopic and/or macroscopic cystic lesions often associated with fibrocystic anomalies in the kidneys. The severity of the renal lesions may overshadow the liver disease, as in the early presentation of autosomal recessive polycystic kidney disease. Conversely, portal hypertension with preserved liver function may dominate a delayed clinical presentation, as in congenital hepatic fibrosis. Cholangitis may develop, especially when the cysts communicate with the biliary system. Malignant change may be a complication.
Embryologically, the hepatobiliary abnormalities are thought to stem from ductal plate maldevelopment in different parts of the biliary tree [1,2].
The ductal plate is a sleeve of epithelium, one and then two cells thick, which forms in the mesenchyme surrounding portal vein branches from bipotential liver progenitor cells—that is cells that may form either hepatocytes or bile duct epithelium. During the development of the liver and biliary system, ductal plates are remodelled into mature tubular ducts which eventually form (in descending size interhepatically from the hilum) hepatic ducts, segmental ducts, area ducts, interlobular ducts and the smallest bile duct branches. Since the various segments of the intrahepatic duct system develop successively throughout fetal life, it is likely that the timing of the inherited malfunction will determine the type and extent of ductal injury [3].
Ductal plate malformation during embryogenesis may also account for a subgroup of biliary atresia, though in most cases this condition is due to necroinflammatory destruction and fibrous obliteration of normally formed extrahepatic bile ducts.
The various types of fibrocystic disease will be described under separate headings (Table 14.1). This classification is based on the dominant clinicopathological features at presentation. However, the different entities are part of a spectrum and may coexist in various combinations [3]. In addition to the influence of the genetic mutations identified so far, environmental factors, in particular superimposed infection, may modulate the disease process. Moreover, the natural history of these disorders has been modified by improved treatment such as renal transplantation and successful management of portal hypertension. These enable patients to live longer but with liver complications or late renal failure.
Table 14.1. Hereditary fibrocystic diseases of the liver: clinical presentation and associated renal disorders
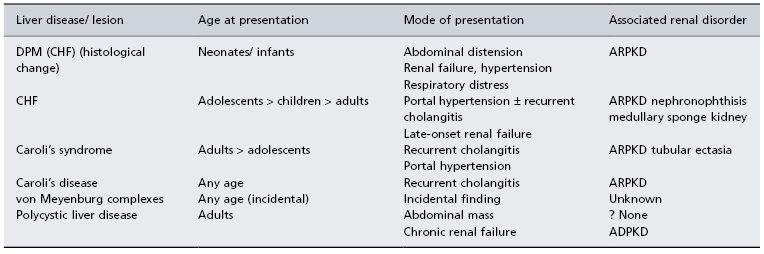
DPM, ductal plate malformation; CHF, congenital hepatic fibrosis; ARPKD, autosomal recessive polycystic kidney disease; ADPKD, autosomal dominant polycystic kidney disease.
Autosomal Recessive Polycystic Kidney Disease
Autosomal recessive polycystic kidney disease (ARPKD) has an estimated incidence of 1/10 000 live births and comprises a spectrum of clinical and histopathological manifestations, the constant features being ductal plate malformation of the intrahepatic bile ducts and fusiform dilatation of renal tubules.
The gene for ARPKD, called polycystic kidney and hepatic disease-1 (PKHD1), has been mapped to chromosome 6 (6p21.1) [4]. It is a large gene (67 exons) encoding the protein fibrocystin [5,6]. This is expressed in renal collecting ducts and bile ducts, but its biological function is unknown [5,6]. More than 100 mutations are known. Most renal patients are compound heterozygotes. There is no clear genotype/ phenotype relationship for predominant renal or hepatic involvement. Mutations leading to truncated transcripts have been described with a severe renal, often lethal, phenotype [7].
Clinical Features
The severity of the renal lesion determines the mode of presentation. In the perinatal or infantile period, rapidly progressive renal failure is associated with large symmetrical renal masses due to radially orientated and fusiform corticomedullary cysts. Infants may be stillborn or die in early infancy from renal failure, fluid retention or bronchopneumonia. The liver lesion is usually clinically silent. The affected children may have cardiac anomalies (ventricular septal defect, pulmonary valve stenosis) and dysmorphic features (‘Potter face’—squashed nose, micrognathia, large low-set ears, epicanthal folds), possibly secondary to severe hydramnios. Cases with a lesser degree of renal involvement present later in life—late childhood or juvenile forms—with progressive renal failure and/or portal hypertension. The condition then merges with congenital hepatic fibrosis (CHF). Often patients are referred to the hepatologist while on chronic renal dialysis because of complications of portal hypertension, such as splenomegaly with hypersplenism and bleeding varices, and/or cholangitis [8,9].
Pathology
Histologically, the liver lesion in ARPKD ranges from a persistent hyperplastic ductal plate with scanty portal fibrosis to marked excess of ductular structures at the periphery of the fibrotic portal tracts. At an early stage, portal fibrosis resembles primitive mesenchyme; later the collagen is more mature and the lesion is indistinguishable from CHF. There are no visible cysts in the liver, which may be enlarged and firm, showing a fine reticular pattern of fibrosis [10].
Imaging
Plain abdomen X-ray, ultrasound and intravenous pyelogram are the most useful investigations in infancy. Bile duct abnormalities can be diagnosed by magnetic resonance cholangiopancreatography (MRCP) [11].
Treatment
For those patients presenting in childhood with gastrointestinal bleeding, endoscopic sclerotherapy or banding of varices is the treatment of choice. Failure to control bleeding with these techniques necessitates portosystemic shunting [12]. Combined liver–kidney transplant is advisable for children with established end-stage renal failure and advanced chronic liver disease, since immunosuppression after isolated renal transplant may increase the frequency cholangitis with worsening liver function.
Adult Polycystic Disease
In adult polycystic disease, liver cysts are probably developmental and are frequently associated with autosomal dominant polycystic kidney disease (PKD). The latter is better understood than the liver disease. At least three different genes appear to be involved.
PKD-1 on chromosome 16p13.3 expresses polycystin 1 which is thought to have a role in epithelial cell differentiation and maturation, and in cell–cell interactions [13,14]. Loss of function of this protein may be one prerequisite for cyst formation but a further somatic event is thought necessary.
PKD-2 on chromosome 4q21–23 expresses polycystin 2. Polycystin 1 and 2 interact through their C-terminal cytoplasmic tails suggesting that they function together through a common signalling pathway. Polycystic kidney disease type 2 is clinically milder than type 1.
Polycystic liver disease is most often associated with autosomal dominant polycystic kidney disease. However, polycystic liver may be an isolated finding and is genetically linked to chromosome 19p13.2–13.1 [15].
The molecular mechanism responsible for cyst formation is not clear. They may arise from failure of supernumerary intrahepatic bile ducts within the hepatic embryonic anlage to involute when the biliary system forms. When the original segment of blind bile ducts is replaced by a second generation of highly active proliferating ducts, redundant ducts may become distorted and form cysts. The second-generation bile ducts are normal so there is no biliary dysfunction.
Pathology
Depending on the number and size of cysts, the liver may be normal or greatly enlarged. Cysts may be scattered diffusely or restricted to one lobe, usually the left. The outer surface may be considerably deformed. A cyst may vary in size from a pin’s head to a child’s head, the largest having a capacity of over 1 litre. They are rarely greater than 10 cm in diameter. The larger ones are probably formed by rupture of septa between adjacent cysts, and the cut liver may display a honeycomb appearance. The cavities are thin walled and contain clear or brown fluid due to altered blood. They never contain bile because they are not in continuity with the biliary tract. They may be complicated by haemorrhage or infection.
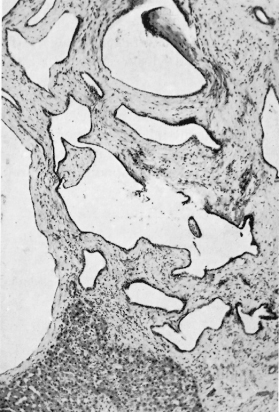
Histologically the lobular architecture is unchanged and liver cells are normal. The cystic areas are related to the bile ducts and to biliary microhamartomas in the portal areas. They are surrounded by a fibrous tissue capsule and lined by columnar or cuboidal epithelium (Fig. 14.3).
Fig. 14.3. Polycystic disease of the liver. The cysts vary in size and are lined by flattened epithelium. (H & E, ×63.)
Frequently, there is cystic disease of other organs, including kidneys, spleen, pancreas, ovary and lungs. About half the patients with polycystic disease of the liver have polycystic kidneys. The majority (50–88%) of patients with polycystic kidneys have a polycystic liver [16]. The prevalence of hepatic cysts increases with age, being approximately 20% in the third decade rising to 75% in the seventh decade.
Cyst Fluid
Fluid has been obtained using needle aspiration under ultrasound guidance [17]. The constituents and response to secretin support the concept that cyst fluid is formed by functioning bile duct epithelium lining the cysts.
Clinical Features
In many patients the liver lesion is diagnosed incidentally during scanning or at autopsy. Sometimes the patient presents with some other disease or with polycystic kidneys.
Patients with symptoms and signs are usually in the fourth or fifth decade. The patient complains of abdominal distension and dull abdominal pain. Pressure on the stomach and duodenum causes epigastric discomfort, nausea, flatulence and occasional vomiting. Acute pain may be due to rupture of, or haemorrhage into, a cyst.
Cysts tend to be larger in women who have been pregnant [18]. Hormone replacement therapy is associated with an approximate 5% enlargement of the liver over a year [19]. There is no increase in symptoms. On the basis of these data hormone replacement therapy is not withheld when clinically indicated [19].
Ascites, obstructive jaundice and hepatic venous outflow obstruction [20] are rare.
On examination the liver may be impalpable or so large that it seems to fill the whole abdomen. The edge is firm and nodules can be palpated. There may be difficulty in distinguishing cysts from other types of liver nodule. The spleen is not enlarged.
Bilaterally enlarged irregular kidneys may suggest associated renal cysts which may be symptomatic.
Hepatic fumction is excellent because the liver cells are preserved. Serum alkaline phosphatase and γ-glutamyl transpeptidase may be increased but bilirubin is normal.
The serum carbohydrate antigen CA 19-9 may be elevated, and levels correlate with high levels found in cyst fluid and polycystic liver volume [21]. Serum levels increase with liver cyst infection in kidney transplant recipients [22].
Portal venous obstruction rarely may result in oesophageal varices which bleed [23].
Imaging
Ultrasound is the most satisfactory method of diagnosis (Fig. 14.4). CT scanning (Fig. 14.5) is also useful in symptomatic patients with multiple cysts to show how much normal liver remains. This helps with planning surgical options.
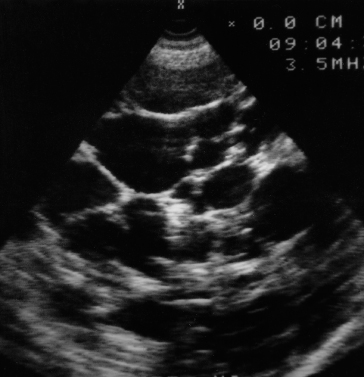
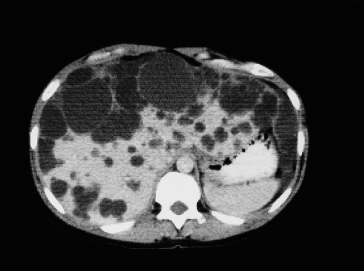
Differential Diagnosis
Polycystic liver should be suspected in an apparently well person, often over 30 years of age, with nodular hepatomegaly, but no evidence of hepatic dysfunction, associated with polycystic kidney or a positive family history.
Polycystic liver may be confused with hydatid disease (Chapter 32).
Metastases are accompanied by malaise, weight loss, rapid increase in size of the liver, and, possibly, evidence of a primary neoplasm.
Cirrhosis may be accompanied by signs of hepatocellular disease and the spleen is usually enlarged.
Prognosis and Treatment
Polycystic disease of the liver is compatible with long life.
The prognosis is determined by the extent of associated renal cystic disease. Carcinoma is very rare. Surgery is rarely necessary and aspiration under ultrasound control is easy and effective in controlling acute symptoms. However, the fluid returns.
In clinical trials somatostatin analogues do reduce liver size (by 3–6%) [24,25] and may reduce perception of pain. This approach holds promise for selected patients but is not yet in clinical practice outside trials.
There are several surgical techniques, the choice depending upon the extent of disease [26]. Patients with a limited number of large cysts may be treated by fenestration which can be performed laparoscopically [27]. Where there is localized involvement of liver parenchyma by multiple medium-sized cysts but with adjacent large areas of normal parenchyma shown by CT, operative fenestration with or without hepatic resection produces symptomatic improvement in most cases [28,29]. In patients with massive diffuse involvement of the majority of liver parenchyma by all sizes of liver cysts with only a small amount of normal parenchyma between them, fenestration may be useful but carries a high morbidity and mortality. In patients with severe limitation of daily activity and failed previous treatment, liver transplantation can be done (combined with kidney transplantation if necessary) and has a 1-year survival of 89% [30].
Successful liver transplantation has been reported using a donor liver with polycystic change [31].
Congenital Hepatic Fibrosis
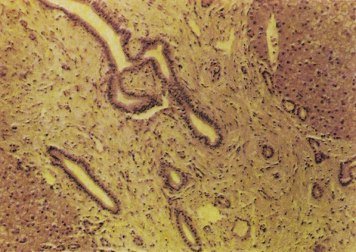
CHF consists histologically of broad, densely collagenous fibrous bands surrounding otherwise normal hepatic lobules (Fig. 14.6). The bands contain large numbers of microscopic, well-formed bile ducts (Fig. 14.7), some containing bile. Arterial branches are normal or hypoplastic, while the veins appear reduced in size. Inflammatory changes are not seen. It may be associated with congenital dilatation of the intrahepatic bile ducts (Caroli’s syndrome).
Fig. 14.6. Congenital hepatic fibrosis. Broad bands of fibrous tissue containing bile ducts separate and surround liver lobules. (Silver impregnation, ×36.)
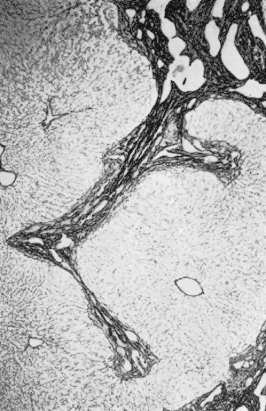
Fig. 14.7. Congenital hepatic fibrosis. Portal area shows dense mature fibrous tissue with a number of abnormal bile ducts. (H & E, ×40.)
The disease appears both sporadically and in a familial form. In the latter, the mode of inheritance is autosomal recessive. A ductal plate malformation of interlobular bile ducts has been suggested as the pathogenetic mechanism [1].
Portal hypertension is common. Occasionally, this may be due to defects in the main portal veins. More often it is caused by hypoplasia or fibrous compression of portal vein radicles in the fibrous bands surrounding the nodules.
Associated renal conditions include renal dysplasia, adult-type polycystic kidneys [32] and nephronophthisis (medullary cystic disease).
Clinical Features
CHF is diagnosed predominantly in children and adolescents, rarely as an isolated entity, more often in association with ARPKD, or less frequently with renal medullary cystic lesions, in particular nephronophthisis and medullary sponge kidneys. The clinical presentation is with signs of portal hypertension (splenomegaly with hypersplenism or variceal haemorrhage) or incidental finding of isolated firm hepatomegaly (Fig. 14.8).
Fig. 14.8. Girl of 8 years with hepatosplenomegaly discovered at routine examination. Liver biopsy showed congenital hepatic fibrosis. Note normal development.
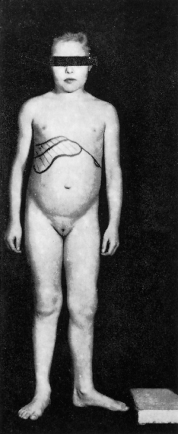
The age at presentation is typically between 5 and 13 years [33]. Sexes are equally affected.
CHF is often misdiagnosed as cirrhosis. An important clue is that hepatocellular function remains essentially normal. A cholangitic form is recognized with subclinical or symptomatic episodes of recurrent cholangitis requiring exclusion of an associated Caroli’s disease. Rarely, recurrent cholangitis may progress to a true biliary cirrhosis with progressive impairment of the liver function. In a few cases the disease manifests in adulthood (latent form) [3].
CHF has been reported in association with familial congenital heart disease [34], pulmonary arteriovenous fistulae [35] and multiple gastric ulcers [36]. A number of malformation syndromes are characterized by hepatic histological changes resembling those of CHF (Table 14.2).
Table 14.2. Malformation syndromes reported with liver histological changes resembling those of congenital hepatic fibrosis
| Meckel’s syndrome: encephalocoele, polydactyly and cystic kidneys [37] |
| Ivemark’s syndrome: dysplasia of the pancreas, liver and kidneys, together with cysts in the pancreas and liver in some cases [38,39] |
| Ellis van Creveld syndrome or chondroectodermal dysplasia [40] |
| Nephronophthisis–congenital hepatic fibrosis syndrome with retinal lesions, mental retardation, cerebellar hypoplasia and osseous abnormalities [41] |
| Jeune syndrome: skeletal dysplasia, pulmonary hypoplasia and retinal lesions [42,43] Laurence–Moon–Biedl syndrome [44] |
Carcinoma, both hepatocellular and cholangiocarcinoma, may be a complication [45,46] as may adenomatous hyperplasia [47].
CHF is a feature of the rare disorder carbohydrate-deficient glycoprotein syndrome type Ib (CDGS 1b) [48], other features including cyclical vomiting, protein-losing enteropathy and prothrombotic tendency. Abnormal glycosylation is caused by phosphomannose isomerase deficiency, which can be demonstrated by transferrin isoelectric focusing (IEF). Regular mannose supplements bypass the enzymatic pathway, correct the biochemical abnormality and improve the clinical symptoms [49]. Despite difficulties in interpreting a link between the biochemical defect in CDGS 1b and the histological features of CHF, screening all patients with CHF for glycosylation defect by transferrin IEF appears justified.
Investigations
Serum protein, bilirubin and transaminase levels are usually normal, while serum alkaline phosphatase values are occasionally increased.
Liver biopsy is essential for diagnosis, but because of the tough consistency of the liver it may be difficult. Liver histology shows bridging fibrous septa, which sharply delineate areas of parenchyma with a normal architecture. The fibrous bands contain numerous ductular structures similar to those seen in infants with ARPKD and it is arguable whether CHF is simply a variant of ARPKD. Some are dilated and may contain bile. The portal vein branches are inconspicuous or reduced to numerous thin radicles. Focal cholangiolitis and copper-associated protein deposition may lead to a misleading diagnosis of cirrhosis.
Ultrasound shows very bright areas of echogenicity due to the dense bands of fibrous tissue. Direct cholangiography in patients with congenital hepatic fibrosis alone shows tapered intrahepatic radicals suggesting fibrosis. MR cholangiography may show duct abnormalities including biliary cysts in some patients. The association of these with CHF has been termed Caroli’s syndrome. Choledochal cysts may also occasionally be seen [50].
Portal venography
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


