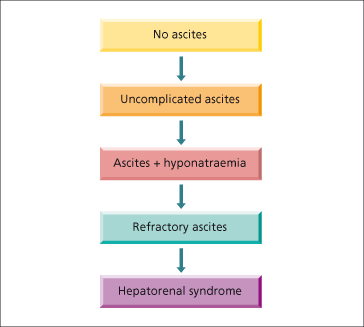
The mechanisms of ascites formation in cirrhosis are complex but portal (sinusoidal) hypertension and renal retention of sodium are universal. The natural history of cirrhotic ascites progresses from diuretic-responsive (uncomplicated) ascites to the development of dilutional hyponatraemia, refractory ascites, and finally, hepatorenal syndrome (HRS) (Fig. 10.2). While 1-year survival in patients who develop ascites is 85%, it decreases to 25% once it has progressed to hyponatraemia, refractory ascites or HRS [4].
Treatment of ascites has not resulted in a significant improvement in survival. However, treating ascites is important, not only because it improves quality of life but because spontaneous bacterial peritonitis (SBP), a lethal complication of cirrhosis, does not occur in the absence of ascites. New treatments are being evaluated that modify its pathophysiology, such as the transjugular intrahepatic portosystemic shunt (TIPS) for refractory ascites and vasoconstrictors for HRS. Liver transplantation is the ultimate therapy and should be considered when the patient first presents with ascites.
Mechanisms of Ascites Formation
In cirrhosis, the source of ascites is mainly the hepatic sinusoids. Therefore sinusoidal hypertension is the initial mechanism that determines leakage of ascites into the peritoneal space [5,6]. Sinusoidal hypertension results from hepatic venous outflow block secondary to regenerative nodules and fibrosis. The other essential factor in the pathogenesis of cirrhotic ascites is sodium and water retention which allows for the replenishment of the intravascular volume and maintenance of ascites formation [7]. Inappropriate sodium retention is either secondary to vascular changes (underfill and peripheral arterial vasodilatation hypotheses) or as a primary event (overfill theory) (Table 10.1).
Table 10.1. Sequence of events for the hypotheses of ascites formation
| Underfill/ peripheral arterial vasodilatation theory | Overfill theory | |
| Primary event | Vascular | Renal |
| Secondary event | Renal | Vascular |
Sinusoidal Hypertension
Similar to gastro-oesophageal varices, in which a minimal portal pressure gradient of 12 mmHg is needed for their presence [8]; the development of ascites also requires a minimal portal pressure gradient of 12 mmHg [5,6]. A threshold portal pressure gradient of 10 mmHg or more has been defined as ‘clinically significant portal hypertension’ because it best predicts the development of complications of cirrhosis, such as ascites [9,10].
Sodium Retention
In patients with cirrhosis and ascites, the normal regulation of sodium balance is lost. Sodium is retained avidly; urinary sodium excretion is often below 5 mmol/day. Inappropriate sodium retention occurs even in the absence of ascites [11].
Vasodilatation Theory (Fig. 10.3)
Arterial vasodilatation, a haemodynamic abnormality typical of the patient with cirrhosis, is the most likely mechanism that explains sodium retention [7]. An increased production of the vasodilator nitric oxide (NO) is considered the main cause of vasodilatation [12]. In experimental models of cirrhosis, inhibition of NO synthase increases systemic blood pressure and renal sodium excretion, resulting in a reduced volume of ascites [13,14]. Other vasodilators implicated in the vasodilatation of cirrhosis include adrenomedullin, carbon monoxide, endocannabinoids, prostacyclin, tumour necrosis factor alpha and urotensin [15].
Fig. 10.3. The peripheral arterial vasodilatation hypothesis for ascites formation in cirrhosis [7].
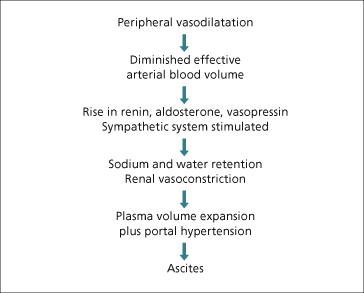
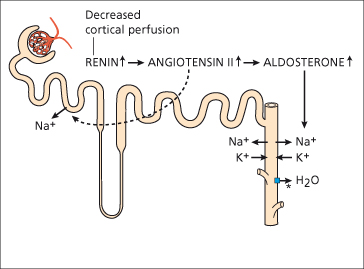
Arterial vasodilatation results in a reduction in ‘effective’ arterial blood volume and a decrease in systemic arterial pressure, leading to the activation of the renin–angiotensin–aldosterone system (RAAS) and the sympathetic nervous system (through carotid sinus baroreceptors). Renin is produced by the kidney (juxtaglomerular apparatus) in response to low blood volume and β-adrenergic stimulation. Under the influence of renin, angiotensinogen (produced by the liver) is converted to angiotensin I (a decapeptide), which in turn is converted to angiotensin II (an octapeptide) by angiotensin-converting enzyme (ACE). Angiotensin II is the main stimulant to the synthesis and secretion of aldosterone, a mineralocorticoid, from the glomerular cells of the adrenal cortex. Aldosterone acts on cells in the collecting duct(ule) and, through a cytoplasmic interaction, increases both luminal uptake and basolateral passage of sodium (Fig. 10.3). Natriuresis after spironolactone, an aldosterone antagonist, supports hyperaldosteronism as a major contributor to sodium retention in cirrhosis [16].
In addition to sodium (and water) retention, angiotensin II is a potent vasoconstrictor (both venules and arterioles), a potent stimulant for the non-osmotic release of antidiuretic hormone (ADH) from the posterior pituitary and a potent activator of the adrenergic system (Fig. 10.4).
Fig. 10.4. Mechanisms of increased sodium and water reabsorption in cirrhosis. * Increased ADH-stimulated water reabsorption in collecting ducts.
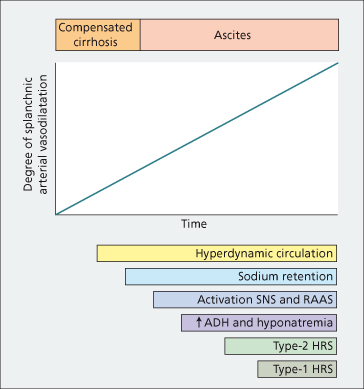
Bacterial translocation to mesenteric lymph nodes with increased endotoxin production and consequent stimulation of cytokine synthesis plays a major role in enhancing vasodilatation in animals with cirrhosis and ascites [17,18]. Further vasodilatation, with further activation of the RAAS, leads to hyponatraemia (through secretion of ADH) [19], and to the HRS (through maximal renal vasoconstriction) [7]. The time course of circulatory, neurohumoral and renal function abnormalities is depicted in Fig. 10.5 [20].
Fig. 10.5. Time course of circulatory, neurohormonal and renal function abnormalities in cirrhosis (in sequence of peripheral arterial vasodilation theory). ADH, antidiuretic hormone; HRS, hepatorenal syndrome; RAAS, renin–angiotensin–aldosterone system; SNS, sympathetic nervous system.
(From [20] with permission.)
Overfill Theory (Fig. 10.6)
The presence of normal or low levels of plasma renin activity in about a third of patients with cirrhosis and ascites, suggests that in some cases sodium retention occurs unrelated to vasodilatation. An alternative proposal is that, early on in the process, there is a primary renal change—responding to hepatic insufficiency or sinusoidal hypertension—that leads to sodium retention (overfill theory). Several signals have been suggested: reduced hepatic synthesis of a natriuretic agent, reduced hepatic clearance of a sodium-retaining hormone, or a ‘hepatorenal reflex’ of unknown aetiology. This theory is based on findings of sodium handling abnormalities, in the absence of systemic vasodilatation or arterial under-filling, when patients with preascitic cirrhosis are challenged with a sodium load [21]. This hypothesis proposes that primary sodium and water retention lead to expansion of the plasma volume, an increase in cardiac output and a fall in systemic vascular resistance (vasodilatation).
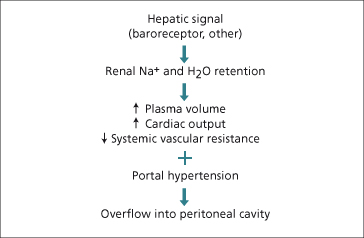
Whether vasodilatation is a primary or secondary event, therapy that counteracts the mechanisms that lead to vasodilatation (e.g. NO inhibition) or vasodilation itself (e.g. vasoconstrictors), improves renal haemodynamics and increases sodium excretion [13,22].
Other Renal Factors
Atrial Natriuretic Peptides (ANP)
The plasma concentration of ANP is markedly increased in patients with cirrhosis and ascites, regardless of plasma levels of renin, aldosterone and noradrenaline (norepinephrine). This is a potent natriuretic peptide released from the cardiac atria, probably in response to intravascular volume expansion. In compensated cirrhosis, ANF may maintain sodium homeostasis despite the presence of mild antinatriuretic factors. In later stages, renal resistance to ANF develops, rendering it ineffective [23]. Therefore, sodium retention in cirrhosis cannot be explained on the basis of a deficient synthesis of natriuretic peptides.
Prostaglandins
Several prostaglandins are synthesized in the kidney and have both vascular and tubular actions. Although they are not primary regulators, they modulate the effects of other factors and hormones locally. Prostaglandin (PG) I2 and E2 are vasodilators, and also increase sodium excretion through vasodilatation and a direct effect on the loop of Henle. They inhibit cyclic adenosine monophosphate (cAMP) synthesis, thereby interfering with the action of vasopressin (ADH). PGI2 is synthesized in the tubules and increases sodium and water excretion. Therefore, prostaglandins have a significant role in sodium and water homeostasis. In conditions where there is a reduced circulating volume, which includes cirrhosis, there is increased prostaglandin synthesis. This counterbalances renal vasoconstriction by antagonizing the local effects of renin, angiotensin II, endothelin 1, vasopressin and catecholamines.
The importance of this role is demonstrated clinically by the renal dysfunction precipitated by the administration non-steroidal anti-inflammatory agents to decompensated [24] and compensated [25] patients with cirrhosis. Without the vasodilatory influence of prostaglandins, renal blood flow and glomerular filtration rate fall because of unopposed vasoconstriction due to renin and other factors. Such an imbalance may be a trigger for HRS.
Circulation of Ascites
Once formed, ascitic fluid can exchange with blood through a large capillary bed under the visceral peritoneum. This plays a vital, dynamic role, sometimes actively facilitating transfer of fluid into ascites and sometimes retarding it. Ascitic fluid is continuously circulating, with about half entering and leaving the peritoneal cavity every hour, there being a rapid transit in both directions. The constituents of the fluid are in dynamic equilibrium with those of the plasma. Rate of ascitic fluid reabsorption is limited to 700–900 mL daily.
Summary (Fig. 10.5)
Ascites in cirrhosis results from sinusoidal hypertension and sodium retention. The most accepted theory for sodium retention is the peripheral arterial vasodilatation hypothesis, which proposes that renal sodium (and water retention) is due to reduced effective blood volume secondary to peripheral arterial vasodilatation (Figs 10.3, 10.4, 10.5). The renal changes are mediated by stimulation of the RAAS, an increase in sympathetic function, and other systemic and local peptide and hormone disturbances. The overfill view suggests that renal retention of sodium is primary with secondary vascular changes and accumulation of ascites and oedema. Depending on the degree of circulatory changes (Table 10.2), these same mechanisms will lead to hyponatraemia and, at the extreme end of severity of renal and vascular changes, HRS develops (Fig. 10.5).
Table 10.2. Circulatory changes in patients with cirrhosis
| Increased | Plasma/ total blood volume |
| Non-central blood volume | |
| Cardiac output | |
| Portal pressure and flow | |
| Reduced | Central blood volume |
| Arterial blood pressure | |
| Splanchnic vascular resistance | |
| Systemic vascular resistance | |
| Renal blood flow |
Clinical Features
Symptoms
The most frequent symptoms are increased abdominal girth (the patient notices tightness of the belt or garments around the waist) and recent weight gain [26]. As fluid continues to accumulate, it leads to elevation of the diaphragm that may cause shortness of breath. Fluid accumulation may also be associated with a feeling of satiety and generalized abdominal pain. The rapid onset of symptoms in a matter of weeks in ascites helps to distinguish it from obesity, which develops over a period of months to years.
Examination
The presence of ascites in patients with cirrhosis denotes a decompensated, more advanced stage of cirrhosis, therefore stigmata of cirrhosis are usually present (spider angiomata, palmar erythema, muscle wasting). There may also be jaundice and signs of portal hypertension, such as splenomegaly and abdominal wall collaterals. Inferior vena caval collaterals result from a secondary, functional block of the inferior vena cava due to pressure of the peritoneal fluid. They commonly run from the groin to the costal margin or flanks and disappear when the ascites is controlled and intra-abdominal pressure is reduced.
Physical examination is relatively insensitive for detecting ascitic fluid, particularly when the amount is small and/or the patient is obese. Patients must have at least 1500 mL of fluid to be detected reliably on physical examination. The clinical diagnosis of ascites will be questionable or incorrect in roughly a third of the cases [27]. When present in small amounts, ascites can be identified by bulging flanks. Flank dullness is very sensitive in detecting ascites [28]. When flank dullness is detected, it is useful to see whether it shifts with rotation of the patient (shifting dullness). This sign is the most sensitive finding (compared to abdominal distension, bulging flanks and fluid wave) [29]. The fluid wave sign has the poorest sensitivity in the diagnosis of peritoneal fluid, even though its specificity is high [26,28,29]. With tense ascites it is difficult to palpate abdominal viscera, but with moderate amounts of fluid the liver or spleen may be ballotted. The presence of a ballotable liver is a good indicator of the presence of ascites [26].
Associated Conditions
Umbilical Hernias.
Increased intra-abdominal pressure favours the development of diastasis recti or hernias in the umbilical, femoral or inguinal regions or through old abdominal incisions. Hernias develop in about 20 % of patients with cirrhosis and ascites (whereas only 3% have hernias without ascites), and may increase to up to 70% in patients with long-standing, recurrent, tense ascites [30]. The main risks of these hernias are rupture [31] and incarceration, the latter complication observed mostly in patients in whom ascites has reduced after paracentesis, peritoneovenous shunt or after transjugular intrahepatic portosystemic shunt [32]. Once ascites is optimally treated, elective hernia repair with permanent mesh is the best treatment for symptomatic hernias, with far less complications than following emergent repair [33].
Hepatic Hydrothorax.
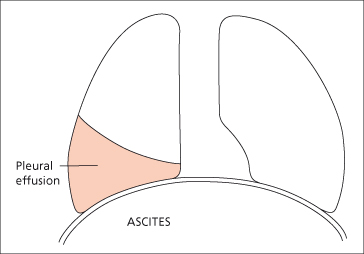
Pleural effusion develops in about 5–10% of patients with cirrhosis [34] and although it usually develops in patients with ascites, hepatic hydrothorax may develop in patients without detectable ascites [35]. Pleural effusion is right-sided in 85%, left-sided in 13% and bilateral in 2% of the cases [36]. It is due to defects in the diaphragm allowing ascites to pass into the pleural cavity (Fig. 10.7). Examination of pleural and ascitic fluid may not be reliable to differentiate an effusion due to local pleural disease from that due to hepatic hydrothorax [37].The diagnosis of hepatic hydrothorax can be established by radionuclide scanning of the chest after the intraperitoneal injection of Tc-99m-labelled sulphur colloid or macroaggregated serum albumin [35]. Presence of radiotracer in the pleural space is demonstrated generally within 2 hours following its intraperitoneal injection [38]. Although large amounts of ascites can accumulate in the peritoneal cavity before resulting in significant patient discomfort, the accumulation of smaller amounts of fluid (1–2 litres) in the pleural space results in severe shortness of breath and hypoxaemia. As pleural fluid is in equilibrium with peritoneal fluid, control depends on medical treatment of ascites. Aspiration is followed by rapid filling up of the pleural space by ascitic fluid. TIPS have been successful [39]; pleurodesis following complete drainage is less successful.
Fig. 10.7. A right-sided pleural effusion may accompany ascites and is related to defects in the diaphragm.
Peripheral Oedema.
This usually follows ascites and is related to hypoproteinaemia. A functional inferior vena caval block due to pressure of the abdominal fluid is an additional factor. The presence of oedema without ascites should therefore lead to investigations of causes of fluid retention other than ascites.
Ascitic Fluid
Diagnostic paracentesis (of about 30 mL) should always be performed in a patient with new-onset ascites, however obvious its cause. In patients with known cirrhotic ascites, diagnostic paracentesis should be performed at every hospital admission and whenever SBP is suspected. Diagnostic paracentesis is a safe procedure with a very low incidence of serious complications, mostly transfusion-requiring haematomas that occur at a rate of 0.2 to 0.9% [40,41].
Fluid appearance is clear, green, straw-coloured or bile-stained. The volume is variable and up to 70 litres have been recorded. A blood-stained fluid indicates malignant disease or a recent paracentesis or an invasive investigation, such as liver biopsy or transhepatic cholangiography.
Ascites total protein and serum-ascites albumin gradient (SAAG) are two inexpensive tests that, taken together, are most useful in determining the source of ascites (Table 10.3). A high (>2.5 g/dL) ascites total protein occurs with peritoneal involvement (malignancy, tuberculosis) due to leakage of high protein mesenteric lymph from obliterated lymphatics and/or from an inflamed peritoneal surface. A high ascites total protein also occurs in cases of postsinusoidal or posthepatic sinusoidal hypertension when sinusoids are normal and protein-rich lymph leaks into the peritoneal cavity [42]. In cirrhosis, an abnormally low protein content of liver lymph has been demonstrated as a result of deposition of fibrous tissue in the sinusoids (‘capillarization of the sinusoid’), which renders the sinusoid less leaky to macromolecules [43]. On the other hand, the SAAG, which involves subtracting ascites fluid albumin concentration from serum albumin, has been shown to correlate with hepatic sinusoidal pressure [44]. A SAAG more than 1.1 g/dL indicates that there is sinusoidal hypertension and that the source of ascites is the hepatic sinusoid as in the case of cirrhosis, heart failure or Budd–Chiari syndrome [45] (Table 10.3).
Table 10.3. Differential diagnosis among the three most common causes of ascites
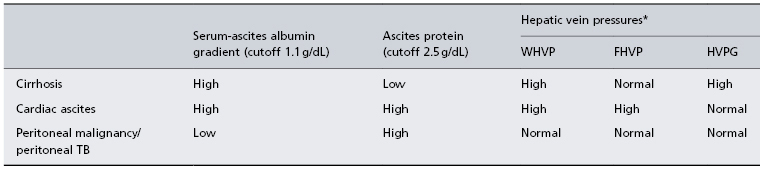
* Only to be performed in equivocal cases. WHVP, wedged hepatic venous pressure; FHVP, free hepatic venous pressure; HVPG, hepatic venous pressure gradient.
Ascites polymorphonuclear cells increase with peritoneal infection or with other intra-abdominal inflammatory conditions such as diverticulitis, cholecystitis. The diagnosis of SBP is established with a polymorphonuclear cell count of more than 250/mm3 [46]. In sterile ascites, ascitic fluid white blood cell count is usually less than 100/mm3 with a predominance of mononuclear cells and a low number of polymorphonuclear cells.
Ascites bacteriological culture is negative in approximately 40% of patients with clinical manifestations suggestive of SBP and increased ascites polymorphonuclear cells [46]. Nevertheless, aerobic and anaerobic cultures should be performed. The percentage of positive cultures increases when ascitic fluid is inoculated directly into blood culture bottles at the bedside, which is the recommended method of culture [46].
Electrolyte concentrations are those of other extracellular fluids.
The rate of accumulation of fluid is variable and depends on the dietary intake of sodium and the ability of the kidneys to excrete it.
Ascitic fluid protein and white cell count, but not polymorph concentration, increase during diuresis.
Radiological Features
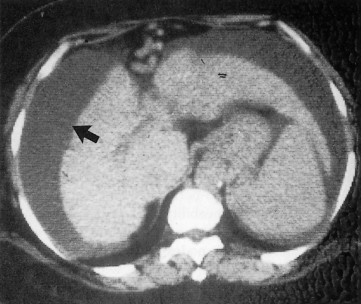
Plain X-ray of the abdomen shows a diffuse ground-glass appearance. Distended loops of bowel simulate intestinal obstruction. Ultrasound and CT scan show a space around the liver and these can be used to demonstrate quite small amounts of fluid (Fig. 10.8).
Differential Diagnosis
Heart Failure/ Constrictive Pericarditis.
Diagnostic points include jugular vein distension and, in constrictive pericarditis, the paradoxical pulse and the radiological demonstration of a calcified pericardium [47]. In both cases SAAG will be more than 1.1 mg/dL and ascites protein will be more than 2.5 g/dL [48]. Right and left heart catheterization and transjugular liver biopsy with measurements of hepatic venous pressure gradient may be necessary to make the differential between cardiac and cirrhotic ascites [27] (Table 10.3).
Malignant Ascites.
There may be symptoms and localizing signs due to the primary tumour. After paracentesis, the liver may be enlarged and nodular. Fluid cytological exam should be performed, although normal endothelial cells in the peritoneum can resemble malignant cells. Massive hepatic metastasis can lead to the development of ascites but since the mechanism of ascites formation is sinusoidal hypertension, these cases of ‘malignant ascites’ will have the characteristics of cirrhotic ascites [49,50].
Tuberculous Ascites.
This should be suspected particularly in the severely malnourished alcoholic who may be febrile. Rarely, lumps of matted omentum can be palpated after paracentesis. Ascitic fluid has many lymphocytes. When suspected, ascites should be stained for tubercle bacilli, and suitable cultures set up.
Mixed Aetiology Ascites.
In patients with mixed ascites (e.g. cirrhosis with superimposed peritoneal malignancy or tuberculosis), the SAAG is high and the ascites protein is low, that is the findings of ascites due to cirrhosis predominate [48,50].
Chylous Ascites.
This results from accumulation of fat, predominantly chylomicrons, in the ascitic fluid. Its appearance is milky and diagnosis is confirmed on a triglyceride ascites content more than 200 mg/dL. The most common cause of chylous ascites is postsurgical disruption of lymphatics. The most common cause of non-surgical chylous ascites is cirrhosis [51,52]. Management is of the underlying cause and a low-fat medium chain triglyceride diet for 3 weeks, or if this fails total parenteral nutrition for 4–6 weeks.
Hepatic Venous Obstruction (Budd–Chiari Syndrome).
This must be considered, especially if the protein content of the ascitic fluid is high and the SAAG is high.
Pancreatic Ascites.
Ascites is rarely gross. It develops as a complication of acute pancreatitis with pseudocyst rupture, or from pancreatic duct disruption. The amylase content of the ascitic fluid is very high.
Ovarian Tumour.
This is suggested by resonance in the flanks. The maximum bulge is anteroposterior and the maximum girth is below the umbilicus.
Spontaneous Bacterial Peritonitis(Table 10.4) [46]
The most common infection in cirrhosis is spontaneous bacterial peritonitis (SBP). It is called spontaneous because it occurs in the absence of a contiguous source of infection (e.g. intestinal perforation, intra-abdominal abscess) and in the absence of an intra-abdominal inflammatory focus (e.g. abscess, acute pancreatitis, cholecystitis). SBP occurs in 9% of hospitalized patients with cirrhosis and accounts for 25% of all infections [53]. It is particularly frequent in severely decompensated cirrhosis. Spontaneous bacterial empyema is an entity akin to SBP in which hepatic hydrothorax becomes infected. Its diagnosis and management are the same as for SBP [54].
Table 10.4. Spontaneous bacterial peritonitis
| Suspect grade B and C cirrhosis with ascites |
| Clinical features may be absent and peripheral WBC normal |
| Ascitic protein usually <1 g/dL |
| Usually monomicrobial and Gram-negative |
| Start antibiotics if ascites >250 mm polymorphs |
| Concomitant albumin use if renal dysfunction or jaundice |
| 20% die |
| 69% recur in 1 year |
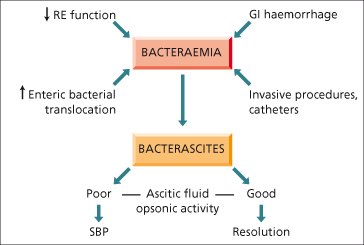
SBP is blood-borne and in 90% monomicrobial. Bacteria of gut origin are the most commonly isolated causative organisms. Therefore, migration of enteric bacteria across the intestinal mucosa to extraintestinal sites and the systemic circulation (bacterial translocation) has been implicated in its pathogenesis [55]. In cirrhosis, an overactive sympathetic nervous system slows gut motility and facilitates bacterial stasis and overgrowth, thereby facilitating bacterial translocation.
Persistence of bacteria in extraintestinal sites is favoured by impaired host defences. In cirrhosis, host defences are abnormal because of portosystemic shunting and impaired reticuloendothelial function. Neutrophils are abnormal in the alcoholic. Decreased synthesis of proteins, such as complement and fibronectin, result in diminished adhesiveness and decreased bacterial phagocytosis [56]. Ascitic fluid favours bacterial growth and deficient ascitic opsonins lead to defective coating of bacteria which are indigestible by polymorphs. The opsonic activity of the ascitic fluid is proportional to protein concentration and SBP is more likely if ascitic fluid protein is less than 1 g/dL [57] (Fig. 10.9). Infection with more than one organism or with fungi is likely to be associated with colonic perforation or dilatation, or any intra-abdominal source of infection (i.e. secondary peritonitis).
Fig. 10.9. The pathogenesis of spontaneous bacterial peritonitis (SBP) in patients with cirrhosis. GI, gastrointestinal; RE, reticuloendothelial.
SBP should be suspected when a patient with cirrhosis deteriorates, particularly with encephalopathy and/or jaundice. Patients with variceal bleeding or with previous SBP are at particular risk. Pyrexia, local abdominal pain and tenderness, and systemic leukocytosis may be noted. These features, however, may be absent and the diagnosis is made following a high index of suspicion with examination of the ascitic fluid.
The diagnosis of SBP is established with an ascites polymorophonuclear count more than 250/ mm3 [46]. The infecting organisms are commonly Escherichia coli or group D streptococci [58]. Anaerobic bacteria are rarely found. Blood cultures are positive in 50%.
Bacterascites (positive culture, polymorphonuclear cells <250/mm3) may resolve without treatment but can progress to SBP [59].
Patients with SBP are particularly at risk of renal complications, probably related to systemic vascular changes secondary to inflammatory response to infection generated by tumour necrosis factor and interleukin 6 [60].
Prognosis
With SBP, 10–20% of patients will die during that hospital admission. The 1-year probability of SBP recurrence is 69% and median survival of a patient who develops SBP is 9 months [61]. Mortality depends on the development of renal dysfunction [62] and the site of acquisition of the infection, with nosocomial infection being an important predictor of death [63–65].
SBP resolution and immediate survival are 100% in community-acquired SBP that is uncomplicated (i.e. no renal dysfunction, no encephalopathy) whether patients receive oral or intravenous antibiotics [66].
Treatment
Antibiotics should be started empirically in all patients with ascites showing more than 250 polymorphonuclear cells/mm3, except in those in whom a local inflammatory reaction is identified (e.g. diverticulitis, cholecystitis, etc.).
Five to seven days of a third-generation cephalosporin such as cefotaxime administered intravenously is usually effective [67,68]. For cefotaxime the optimal cost-effective dosage is 2 g every 12 h. Amoxycillin-clavulanic acid is as effective as cefotaxime [69].
The antibiotic choice should be reviewed once results of ascitic culture and sensitivity of the bacterial isolates are known. Because of renal toxicity, aminoglycosides should be avoided.
In a randomized study the administration of intravenous albumin to patients with SBP treated with cefotaxime significantly reduced the incidence of renal impairment (10 vs. 33%) and hospital mortality (10 vs. 29%) [62]. Patients that benefit most from the use of albumin are those with renal dysfunction at baseline (creatinine >1.0 mg/dL and/or blood urea nitrogen >30 mg/dL) and serum bilirubin more than 4 mg/dL [62,70,71].
Success rates for cefotaxime and amoxicillin-clavulanic may be as low as 44% in nosocomial SBP because of the presence of multidrug-resistant organisms [72,73]. Extended spectrum antibiotics (e.g. carbapenems, piperacillin/ tazobactam) should be used as initial empirical therapy in patients with hospital-acquired SBP, particularly in those who had been on beta-lactams during admission, had been recently hospitalized or on quinolone prophylaxis [72].
Secondary bacterial peritonitis should be suspected when a suspected SBP fails to respond to antibiotic therapy.
Because of reduced survival, SBP is an indication to consider hepatic transplantation, particularly if recurrent.
Prophylaxis
Long-term prophylaxis will lead to the emergence of resistant bacteria [53]. Therefore, only patients at the highest risk of developing SBP should receive antibiotic prophylaxis.
The risk of SBP is particularly high in patients with cirrhosis with upper gastrointestinal haemorrhage. Oral administration of norfloxacin (400 mg/12 h for a minimum of 7 days) is currently recommended for this group [46]. However, intravenous ceftriaxone should be considered in high quinolone resistance settings or in patients with two or more of the following: malnutrition, ascites, encephalopathy or serum bilirubin more than 3 mg/dL [74]. SBP and other infections should be ruled out by bacterial cultures before starting prophylaxis.
In patients with a previous episode of SBP, the risk of recurrence during the subsequent year is 40–70%. Oral administration of norfloxacin (400 mg/day) is recommended in such patients, who should then be evaluated for liver transplantation [46,75].
There is currently insufficient evidence to recommend prophylaxis for patients with a low ascitic fluid protein (<1 g/dL). However, norfloxacin prophylaxis appears justified in patients with advanced liver failure (Child–Pugh score >9 points with serum bilirubin level >3 mg/dL) or impaired renal function (serum creatinine level >1.2 mg/dL, blood urea nitrogen level >25 mg/dL, or serum sodium level <130 mEq/L). In these patients, the 1-year probability of first SBP is 60% and is significantly reduced with norfloxacin [76] .
In patients with a high ascitic fluid protein (>1 g/dL) without a past history of SBP, prophylaxis is not necessary as the 1-year probability of SBP is nil [77].
Treatment of Cirrhotic Ascites [78,79]
Therapy of ascites, whether by diuretics or paracentesis, reduces clinical symptoms and improves quality of life. However, although the initial clinical response may be excellent, if fluid loss is excessive it may lead to hyponatraemia, hyperkalaemia, renal failure or encephalopathy. Treatment must therefore be appropriate to the clinical state and the response properly monitored. The approach must be tailored to the patient. The spectrum of therapeutic intervention ranges from sodium restriction alone (rarely used), to diuretic use, therapeutic paracentesis (Table 10.5), and, for the most severe groups, TIPS and eventually liver transplantation.
Table 10.5. General management of ascites
| Diagnostic paracentesis with first presentation or with any symptom/ sign suggestive of SBP |
| 70–90 mmol sodium diet; weigh daily; check serum creatinine and electrolytes |
| Spironolactone 100 mg daily |
| If tense ascites consider paracentesis (see Table 10.7) |
| After 4 days consider adding frusemide (furosemide) 40 mg daily; check serum creatinine and electrolytes |
| Maximum daily weight loss 0.5 kg/day (1.0 kg/day in those with peripheral oedema) |
| Stop diuretics if precoma (‘flap’), hypokalaemia, azotaemia or alkalosis |
| Continue to monitor weight; increase diuretics as necessary |
| Avoid non-steroidal anti-inflammatory drugs |
SBP, spontaneous bacterial peritonitis.
Indications for treatment include the following:
- Symptomatic ascites with abdominal distension sufficient to be obvious and produce physical or emotional distress requires treatment with sodium restriction and diuretics. The presence of subclinical ascites (that seen only on ultrasound without clinical symptoms) may not require active treatment, although to prevent deterioration advice on a reduction in sodium intake is wise. Inappropriate introduction of excessive treatment for ascites may lead to symptomatic hypotension, muscle cramps, dehydration, and renal dysfunction.
- Large ascites, causing abdominal discomfort or pain and/or dyspnoea most often demands paracentesis.
- Tense ascites with pain may lead to eversion and ulceration of an umbilical hernia, which is near to rupture. This complication has a very high mortality, due to shock, renal failure and sepsis, and urgent paracentesis is indicated.
Monitoring during treatment is mandatory. The patient should be weighed daily as it provides a satisfactory guide to progress. Urinary electrolyte (sodium, potassium) determinations are helpful in determining dosage, monitoring the response and assessing compliance. Serum electrolytes and creatinine should be measured two to three times per week while the patient is in hospital. Where liver disease is due to alcohol, the patient should be encouraged to abstain. The mild case is managed as an out-patient by diet and diuretics, but if admitted to hospital, paracentesis is usually a first procedure. In a survey of European hepatologists, 50% used paracentesis initially, followed by diuretics [80]. Fifty per cent regarded complete control of ascites as desirable, whereas the other half was satisfied with symptomatic relief without removing all the ascites. Thus consensus on standardized treatment regimes is difficult to reach because of the clinical spectrum of ascites, the clinical success of the different regimens and the lack of evidence-based studies comparing individual approaches.
Bed rest used to be a feature of initial therapy. Evidence for benefit is sparse but as part of an overall strategy in combination with diuretics it has been found to be beneficial [81]. This may be related to increased renal perfusion and portal venous blood flow during recumbency.
Sodium Restriction/ Diet
The patient with cirrhosis who is accumulating ascites on an unrestricted sodium intake excretes less than 10 mmol (approximately 0.2 g) sodium daily in the urine. Extrarenal loss is about 0.5 g. Sodium taken in excess of 0.75 g will result in ascites, with every gram retaining 200 mL of fluid. Historically, such patients were recommended a diet containing 22–40 mmol/day of sodium (approximately 0.5–1.0 g/day). However, such diet is unpalatable and also compromises protein and calorie intake, which in patients with cirrhosis is critical for proper nutrition. Current recommendations are to use a ‘no added salt’ diet (approximately 70–90 mmol or approximately 1.5–2.0 g/day) combined with diuretics to increase urinary sodium excretion (Table 10.6). In this diet, salt should not be used at the table or when cooking. Also, various foods containing sodium should be restricted or avoided (Table 10.6). Many low-sodium foods are now available.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


