Fig. 5.1
Representative light microscopy showing thickened glomerular basement membranes with spikes and pinholes (arrows) on higher power. (a) PAS stain, 40×; (b) PAS 60×; (c) Silver stain 60×; (d) Trichrome stain 60×
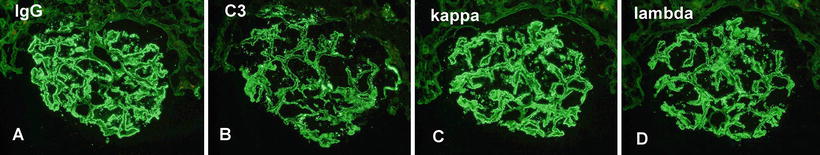
Fig. 5.2
Immunofluorescence microscopy showing (a) granular IgG, (b) C3, (c) kappa light chains, and (d) lambda light chains along the capillary walls
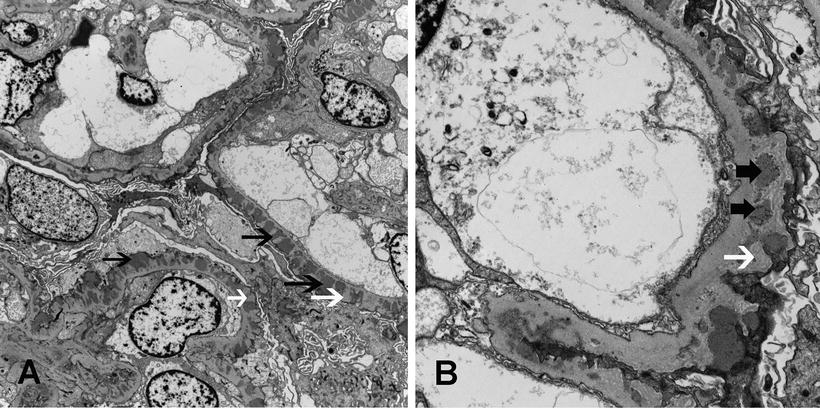
Fig. 5.3
Electron microscopy showing subepithelial electron dense deposits (black arrows) and basement membrane spikes (white arrows) separating the deposits. (a) Membranous nephropathy stage II, with spikes separating the deposits (×4730), and (b) Membranous nephropathy, stage III, with intramembranous deposits (thick black arrows) (×15100)
From the pathology standpoint, it is important to determine whether the membranous glomerulopathy may be due to a secondary cause such as an autoimmune disease, neoplasia, infection, or drugs. Although it is often difficult to determine whether the MN is primary or secondary, there are some features that are helpful in identifying a secondary cause. Features in favor of an secondary cause, in particular an autoimmune disease, include (1) proliferative features—mesangial or endocapillary, (2) full-house pattern of Ig staining including staining for C1q on immunofluorescence microscopy, (3) electron dense deposits in the subendothelial location of the capillary wall and mesangium or along the tubular basement membrane and vessel walls, and (4) endothelial tubuloreticular inclusions on electron microscopy [47]. Electron microscopy showing only few superficial scattered subepithelial deposits may suggest a drug-associated secondary MN. Furthermore, the location of the subepithelial deposits, i.e., subepithelial (homogenous) with subgroups superficial and deep versus subepithelial and intramembranous (heterogenous) deposits should be mentioned since the homogenous group with superficial deposits in one study has been shown to have a better prognosis than the homogenous group with deep deposits and the heterogenous group [48].
Finally, there is also diagnostic value in staining for IgG subclasses in MN that helps differentiate between primary MN and secondary MN due to lupus. IgG1, IgG2, and IgG3 tend to be highly expressed in lupus MN, while IgG1 and IgG4 tend to be highly expressed in primary MN [49].
Natural History
The natural course of MN is variable. Some patients achieve a spontaneous remission of proteinuria and usually maintain a normal renal function over the time; others have a persistent proteinuria without progression, and the remaining patients progress, usually slowly, to ESRD or die from complication related to the nephrotic syndrome. The proportions of these different outcomes are difficult to assess because most of the available data come from studies which are short term and which include not only patients with idiopathic and secondary MN but also treated and untreated patients, or nephrotic and non-nephrotic patients. As a consequence, it is not surprising that the reported outcomes are variable and that some authors consider MN as a relatively benign disease and feel that immunosuppressive agents should be avoided [50], while others stress the potentially poor outcome of MN and suggest an early specific treatment at least in nephrotic patients [51].
For example, Schieppati et al. reported a 72 % renal survival at 8 years for 100 untreated patients with MN [50]. However, in this study, 37 % of patients were non-nephrotic (56 % of patients had proteinuria <5 g/24 h), the median follow-up was only 39 months, and deaths were excluded from the analysis. Despite these exceptions and the “benign” presentation characteristics, 25 % of the patients reached end-stage renal disease (ESRD) by the end of 8 years [50].
Instead, the natural course of nephrotic patients is less favorable. Some 20 % of them may obtain a complete remission of proteinuria usually within 3–5 years [50–54]. However, a review of the papers reporting the long-term outcome of untreated nephrotic patients with MN found that 40–50 % of patients either died or progressed to end-stage renal failure 10–14 years after the clinical onset [55]. This can be illustrated by a recent paper reporting spontaneous remission developing in 32 % of the patients [56]. These authors suggested that patients that were treated conservatively and who did not go into spontaneous remission had final creatinine = 2.4 ± 2.2 mg/dl and eGFR 53 ± 35 ml/min. However, taking into consideration that the mean age at presentation was 51 years and the mean follow-up was 69 months (making these patients ~56 years at follow-up), the recalculated eGFR should be ~30 ml/min if males and ~22 ml/min if females, indicating that a significant loss of kidney function occurred. The paper further underestimates chronic kidney disease because their endpoint was restricted to the absence of chronic dialysis or need for renal transplantation. However, if one uses a different parameter, i.e., loss of kidney function on the basis of GFR, their disease trajectory would indicate a significantly higher incidence of ESRD over time.
Thus, it should not come as a surprise that until recently both in the USA and Europe, MN remained the second or third leading cause of ESRD among the primary glomerulonephritis types [57]. Even patients who do not progress but remain nephrotic are at an increased risk for life-threatening thromboembolic and cardiovascular events. However, if the natural course of the disease is probably worse than generally estimated, current recommendations for immunosuppression have improved outcomes in patients at risk of progression [58].
The progression of the disease is usually slow. However, in few cases, a rapid change in either the degree of proteinuria or in the rate of loss of renal function may occur. This event should raise the possibility of a superimposed condition, e.g., acute renal vein thrombosis, acute interstitial nephritis, or superimposed crescentic glomerulonephritis (ANCA-associated vasculitis or anti-GBM disease).
Predicting Factors:
Evaluating the prognosis is critical in making the decision regarding when and what to use in terms of treatment, e.g., conservative versus immunosuppressive treatment in patients with MN [59–61]. An accurate predictor of outcome of patients with idiopathic MN would allow the separation of those patients who are likely to have a long-term renal survival from those who are likely to progress. This would allow us to target immunosuppressive treatment to patients at high risk of renal disease progression. However, finding useful markers that predict this last group has been difficult. Pathology itself is not helpful in establishing prognosis or predicting response to immunosuppressive therapy, although the combination of severe interstitial fibrosis, vascular sclerosis, and focal glomerular sclerosis is usually associated with a poor renal outcome [62]. Urinary excretion ratios of α[alpha]1-microglobulin, β[beta]2-microglobulin, IgM, and IgG have all been found helpful in assessing the severity of the overall renal injury and to predict outcome in MN [63–68]. Unfortunately quantification of urinary α[alpha]1-microglobulin, β[beta]2-microglobulin, IgM, and IgG is not widely available and thus limits their clinical use. Thus far, the best model for the identification of patients at risk was developed with data derived from the Toronto Glomerulonephritis Registry [69, 70]. This model takes into consideration the initial creatinine clearance (CrCl), the slope of the CrCl, and the lowest level of proteinuria during a 6-month observation period. This risk score assessment has good performance characteristics and has been validated in two geographically diverse MN populations, one from Italy and the other from Finland [70]. Based on this model, patients who present with a normal CrCl, proteinuria ≤4 g/24 h, and stable renal function over a 6-month observation period have an excellent long-term prognosis and are classified as at low risk of progression. Patients with normal renal function and whose CrCl remains unchanged during 6 months of observation, but continue to have proteinuria >4 g but <8 g/24 h, have a 55 % probability of developing chronic renal insufficiency and are classified as medium risk of progression, and patients with persistent proteinuria >8 g/24 h, independent of the degree of renal dysfunction, have a 66–80 % probability of progression to chronic renal failure within 10 years and are classified in the high risk of progression category [59]. Patients with MN who were never nephrotic or who achieved a complete remission of proteinuria have an excellent long-term renal survival. Even a partial remission has been recognized as a predictor of long-term positive outcome in patients with MN [71, 72]. Troyanov et al. reported data on 350 patients with MN and nephrotic syndrome and found that the 10-year renal survival was 100 % in the complete remission, 90 % in the partial remission, and 45 % in the no remission group [71]. Patients in complete or partial remission have similar rate of decline: −1.5 ml/min/year in the complete remission group and −2 ml/min/year in the partial remission group. In contrast, the no remission group lost GFR at a rate of −10 ml/min/year.
Therapy
Symptomatic Treatment
Initial therapy should be supportive and involves restricting dietary protein intake, controlling blood pressure, hyperlipidemia, and edema. In the Modification of Diet in Renal Disease (MDRD) study, patients with proteinuria >1 g/day had a significantly better outcome if their blood pressure was reduced to 125/75 mmHg [73]. Thus, in patients with proteinuric renal disease, the current target for blood pressure control is ≤125/75 mmHg. Reducing protein intake to about 0.6–0.8 g/kg ideal body weight per day also decreases nephrotic range proteinuria [74]. ACEi and/or ARBs are effective antihypertensive agents that can reduce proteinuria and slow progression of renal disease in both diabetic and nondiabetic chronic nephropathy patients, and for these reasons they are the preferred agents to treat hypertension in proteinuric renal diseases. However, evidence that such therapy is beneficial in MN is weak and largely inferential, and the following issues need to be considered: (1) the degree of renal protection is related to the degree of proteinuria reduction and if proteinuria is not lowered, the benefit is substantially attenuated [75, 76]. In the RENAAL trial the renal protective effect of angiotensin II blockade in patients with diabetic nephropathy was nearly fully explained by its anti-proteinuric effect [77]. (2) In patients with MN, the anti-proteinuric effect is modest (<30 % decrease) and is more significant in patients with lower levels of proteinuria [78–80]. (3) Thus, in contrast to diabetic renal diseases, ACEi may not offer the same degree of renal protection to patients with MN [81]. In fact, studies by du Buf-Vereijken et al. [82] and in a review by Troyanov et al. [71], the use of ACEi or ARBs by multivariate analysis did not show an independent value in determining the prognosis of patients with MN. Similarly, Praga et al. showed additional evidence that in patients with nephrotic syndrome (the majority with MN), ACEi were ineffective in reducing proteinuria and that this response in MN patients was associated with a poor renal function outcome [83, 84]. It is also difficult to handle hypercholesterolemia in patients with nephrotic syndrome. Dietary restriction is of little benefit and use of statins can usually obtain a reduction of 25–45 % from baseline values of serum cholesterol.
At any rate, although it is difficult to obtain a good reduction of proteinuria and normalization of hypercholesterolemia even with huge doses of ACE inhibitors, angiotensin receptor blockers, and statins, the current practice is to maximize the doses of these drugs in nephrotic patients. Loop diuretics are usually effective in solving the edema. In difficult cases a thiazide may be added to increase the diuretic activity. Anticoagulation is recommended in patients with severe hypoalbuminemia (<2 g/dl) and in those who already experienced thrombotic events and in bedridden or obese patients [85].
As discussed, the prognosis of patients with non-nephrotic proteinuria is usually excellent, and the conservative symptomatic approach described above should suffice. Obviously these patients should be monitored to ensure that the disease is not worsening since a significant number of patients who present with sub-nephrotic range proteinuria will progress to full nephrotic syndrome within 1–2 years from presentation.
Specific Treatments of Patients with Normal Renal Function
Corticosteroids
Four prospective randomized trials of corticosteroids therapy for MN have been published between 1970 and 1990. In two of these studies, prednisone was administered at low doses for 6 months, and in the other two studies, steroid was given at high doses for 8 weeks.
Black et al. randomly assigned 19 patients with MN and nephrotic syndrome either to symptomatic therapy or to 20–30 mg per day of prednisone for 6 months. After 2 years, 20 % of controls and 40 % of treated patients had daily proteinuria lower than 1 g, but the difference was not significant and side effects of treatment were frequent. The conclusion of the study was that the risks of therapy outweighed the possible benefits [86]. A more solid 6-month therapeutic protocol was evaluated in a Canadian study, in which 158 patients with or without nephrotic syndrome were assigned to receive symptomatic therapy or 45 mg/m2 of prednisone on alternate days. After a mean follow-up of 4 years, there were no differences in remission rates or renal function between the two groups [87]. A problem with this study was that untreated controls had such a benign course that it would have been difficult to find any significant difference even if treatment was effective. In a controlled US Collaborative Study, patients assigned to treatment with prednisone 125 mg every other day for 2 months obtained a higher, but transient, number of remissions (22 vs. 11) and a slower average rate of decline in GFR (−2 % per year vs. −10 % per year) when compared with untreated controls [88]. Two major points of criticism of this study were the short follow-up period and the poorer than expected outcome in the placebo group. A few years later a British Medical Research Trial evaluated the same therapeutic protocol on 107 patients with nephrotic syndrome followed for 3 years [89]. This study was unable to demonstrate any difference between the two groups at any time in either the mean proteinuria or mean plasma creatinine.
Taking together, these controlled trials did not show a benefit of corticosteroids in patients with MN. The lack of response has been confirmed by a meta-analysis published in 1995, which did not find any difference either in the probability of remission (odds ratio 0.97) or in the 5-year renal survival (80 % for both groups) between treated and untreated patients [90]. In addition, a recently published Cochrane meta-analysis showed that, in MN, corticosteroids compared with symptomatic therapy had no beneficial effect on total mortality or ESRD [91]. However, it is important to realize that in these studies, prednisone was given in limited dosage or during a limited period. Thus, one cannot completely exclude that corticosteroids may be of some benefit when using different schedules. Actually, corticosteroids may regulate the actin cytoskeleton of podocytes and their foot processes so improving the permselectivity of the glomerular barrier [92].
Alkylating Agents
A few randomized trials evaluated the role of alkylating agents. Donadio et al. reported on 22 patients randomly assigned to receive oral cyclophosphamide, 1.5–2.5 mg/kg/day for 12 months, or no specific therapy [93]. At the end of treatment, there was a trend to a greater decline of proteinuria in treated patients than in controls (mean reduction 4.7 g per day vs. 2.6 g per day, respectively), but the difference was not significant, and cyclophosphamide was considered ineffective. In a trial including several forms of primary glomerulonephritis, Lagrue et al. randomly allocated 41 nephrotic patients with MN either to chlorambucil (0.2 mg/kg/day for 6 months, then 0.1 mg/kg/day for a further 6 months) or to azathioprine (3 mg/kg/day for 6 months, then 2 mg/kg/day for a further 6 months) or to placebo [94]. At the end of treatment period, chlorambucil-treated patients showed a significant reduction of proteinuria, while no changes were observed in patients given azathioprine or placebo. One year after the treatment period was completed, 81 % of patients assigned to chlorambucil were in complete or partial remission, compared with 9 % of patients treated with azathioprine and 21 % of patients assigned to placebo. However, the prolonged use of chlorambucil was loaded by severe complications, including neoplasia. Two Australian trials investigated the role of cyclophosphamide associated with anticoagulant and antiplatelet agents. Tiller et al. randomly allocated 54 patients to symptomatic therapy or to cyclophosphamide plus warfarin and dipyridamole for 36 months [95]. Patients who completed the treatment obtained a significantly lower proteinuria and higher serum albumin when compared with untreated controls. However, many patients had to stop therapy because of complications. Murphy et al. randomized 40 patients either to symptomatic therapy or to cyclophosphamide for 6 months plus dipyridamole and warfarin for 2 years [96]. Treatment was well tolerated. Compared with controls, treated patients showed a greater reduction of proteinuria and a higher incidence of complete remission compared with controls (9/13 vs. 4/13, respectively).
These controlled trials would show that cytotoxic agents may reduce proteinuria and can improve the chances of remission. The follow-ups of the available studies were too short, however, to evaluate the impact of these agents on renal function and their potential long-term morbidity. Good results have also been reported in noncontrolled studies by using 6–12-month courses of cyclophosphamide and corticosteroids [97].
Alternating Corticosteroids and Alkylating Agents
An Italian multicenter controlled trial evaluated whether giving alkylating agents and steroids in an alternate fashion could achieve therapeutic results while sparing the side effects of these agents. The treatment consisted of three consecutive cycles of 2-month therapy. Each cycle began with an intravenous administration of methylprednisolone, 1 g intravenously daily for 3 consecutive days, followed by oral prednisolone 0.5 mg/kg daily for the rest of the month. Then steroid was stopped and replaced by chlorambucil 0.2 mg/kg/day for 1 month. The 2-month cycle was repeated three times until a total of 6 months of therapy had been given (Table 5.1). Eighty-one adult patients with MN and nephrotic syndrome were randomized to receive either the combined therapy or the symptomatic treatment. The two groups were homogeneous at randomization. The early results of this study were published in 1984 [98], but patients continued to be followed up to 10 years [51]. The probability of having a remission of the nephrotic syndrome, either complete or partial, was significantly higher in treated patients than in untreated (83 % vs. 38 %, respectively). At 10 years, 40 % of treated patients compared with only 5 % of untreated controls were still in complete remission; the slope of the reciprocal of plasma creatinine with time reduced from 1.0 to 0.51 in controls and from 1.0 to 0.84 in treated patients; the probability of surviving without developing ESRD was 92 % in patients given the combined therapy versus 60 % in controls. One patient in the treated group died of lung cancer a few months after randomization and two patients in the control group died, respectively, of cardiac infarct and of hepatorenal failure. Four patients had to stop treatment because of peptic ulcer (two cases), gastric intolerance, and pneumonia. In the long-term, one treated patient became obese and another developed diabetes.
Table 5.1
Therapeutic protocol used in the three Italian multicenter controlled studies
Methylprednisolone + chlorambucil versus symptomatic therapy | |
|---|---|
Months 1, 3, 5 | Methylprednisolone 1 g IV for 3 days, followed by oral prednisone 0.5 mg/kg/day for 27 days |
Months 2, 4, 6 | Chlorambucil 0.2 mg/kg/day for 30 days |
Methylprednisolone + chlorambucil versus methylprednisolone alone | |
Months 1, 3, 5 | Methylprednisolone 1 g IV for 3 days, followed by oral prednisone 0.5 mg/kg/day for 27 days |
Months 2, 4, 6 | Chlorambucil 0.2 mg/kg/day for 30 days |
Months 1, 3, 5 | Methylprednisolone 1 g IV for 3 days, followed by oral prednisone 0.5 mg/kg/48 h for 27 days |
Months 2, 4, 6 | Oral prednisone 0.5 mg/kg/48 h for 30 days |
Methylprednisolone + chlorambucil versus methylprednisolone + cyclophosphamide | |
Months 1, 3, 5 | Methylprednisolone 1 g IV for 3 days, followed by oral prednisone 0.5 mg/kg/day for 27 days |
Months 2, 4, 6 | Chlorambucil 0.2 mg/kg/day for 30 days |
Months 1, 3, 5 | Methylprednisolone 1 g IV for 3 days, followed by oral prednisone 0.5 mg/kg/day for 27 days |
Months 2, 4, 6 | Cyclophosphamide 2.5 mg/kg/day for 30 days |
Another Italian multicenter controlled study compared the combined cyclical combined therapy with methylprednisolone alone given for 6 months at the same cumulative dosage [99] (Table 5.1). At the end of a mean follow-up of 54 months, 64 % of patients given combined therapy versus 38 % of patients given steroids alone were without nephrotic syndrome. There was also a trend toward a better slope of the reciprocal of plasma creatinine in patients given the combined therapy, but the difference was not significant. One patient per group died. Four patients in the group assigned to the combined therapy had severe side effects that reversed completely after treatment was stopped (two cases of pneumonia, one of liver dysfunction, and one of gastric intolerance to chlorambucil). One patient assigned to steroids alone stopped therapy because of pulmonary embolism. A third Italian controlled study compared the effects of the regimen based on steroids and chlorambucil with those of a regimen using the same schedule of steroids but giving cyclophosphamide (2.5 mg/kg/day) instead of chlorambucil [100] (Table 5.1). Of 43 patients assigned to methylprednisolone and cyclophosphamide, 40 (93 %) entered complete or partial remission of the nephrotic syndrome versus 36 of 44 (82 %) for those assigned to methylprednisolone plus chlorambucil. In both groups the reciprocal of plasma creatinine remained unchanged in patients followed up for 2 and 3 years when compared with baseline. Six patients in the chlorambucil group developed severe side effects (two leukopenia, two pneumonia, one anemia and thrombocytopenia, and one nausea). Two patients stopped cyclophosphamide, one because of nausea and one because of a transient ischemic attack. All side effects reversed after treatment was stopped.
Pooling the results of these 3 Italian studies, 174 patients with MN and nephrotic syndrome received a 6-month therapy with corticosteroids alternated with chlorambucil (131 patients) or cyclophosphamide (43 patients). Of them, 72 (41.3 %) obtained complete remission and 72 (41.3 %) had a partial remission as a first event. After a mean follow-up of 54 months, 74 % of patients were in complete (34 %) or partial (41 %) remission. Four patients (2.4 %) progressed to ESRD and two died. Sixteen patients (9 %) suffered from severe side effects that completely reversed after stopping therapy. The results of these trials have been confirmed by an Indian randomized, controlled trial on 93 patients [101]. Of the 47 patients treated with a 6-month course of alternating prednisolone and cyclophosphamide, 34 achieved remission (15 complete and 19 partial remission), compared with 16 (5 complete, 11 partial remission) of 46 in the control group treated with symptomatic therapy. The 10-year dialysis-free survival was 89 % and 65 %, and the likelihood of survival without death, ESRD, and doubling of serum creatinine were 79 % in the treated versus 44 % in the control group. The incidence of infections was similar in the two groups [101]. The long-term studies by Jha and Ponticelli clearly showed that the combined regimen may protect the long-term renal function, with treatment being well tolerated in the majority of patients [51, 101].
Since the most frequent side effects of alkylating agents are leukopenia and infection, we suggest that blood cell count should be checked at least every 7–10 days. The dose of chlorambucil or cyclophosphamide must be halved when leukocytes fall below 5,000 and should be stopped if they fall below 3,000/mm3. Moreover, both chlorambucil and cyclophosphamide also can cause azoospermia; thus young males should be encouraged to deposit their semen in a sperm bank before starting therapy. A main concern with the use of cytotoxic drugs is the potential risk of neoplasia in the long term. Leukemia and, more rarely, lymphoma [102, 103] are the most frequent types of cancer that may develop in patients given chlorambucil, whereas bladder cancer and lymphoreticular tumors [104, 105] are the most frequent malignancies in patients treated with cyclophosphamide. These severe complications appear to be related to the cumulative doses and to the duration of treatment [106–108]. No case of hematological neoplasia has been reported in patients treated with a cumulative dose of chlorambucil of less than 1 g or for a cumulative period shorter than 6 months, and bladder complications are exceptional in patients given cyclophosphamide at doses of 2–3 mg/kg per day for not more than 12 weeks. In the Italian trials none of the patients enrolled developed leukemia, lymphoma, or bladder cancer. Three patients were recognized to have a cancer only after randomization. In order to evaluate whether these cancers represented a chance event or a complication of treatment, the cumulative risk of cancer in patients given methylprednisolone and chlorambucil for 6 months was compared with that of a general white population [100]. Considering together the patients who received methylprednisolone and chlorambucil in the three Italian trials, it was possible to collect information of 662 patient/years. In this population the cumulative risk of developing cancer was 4.53/1,000 patients per year (95 % CI, 0.935–13.241), which is concordant with the average annual incidence of primary cancer of 4.33/1,000 for men and 3.40/1,000 for women among white general population [109].
In a recent study, Hownan et al., randomized 108 patient with progressive MN (defined as a 20 % decline in eGFR before study entry), to treatment with either alternating chlorambucil and corticosteroids for 6 months, cyclosporine monotherapy for 12 months, or supportive care only [110]. The risk of reaching the primary endpoint (a further 20 % decline in eGFR) was significantly lower in the chlorambucil group versus the cyclosporine or supportive care group. Although this study provides support for the use of cyclic corticosteroids/chlorambucil treatment in patients with renal function deterioration, the high progression rate (58 %) in chlorambucil-treated patients, as compared to the low progression rates of 5–8 % in the early randomized controlled trials, may suggest that starting treatment once a significant degree of renal injury had occur may be less effective. It could also be argued that the surrogate renal endpoint used, i.e., a mere 20 % change of eGFR, is inadequate. Too many variables, i.e., lowering of blood pressure, use of diuretics, and change in kidney creatinine handling during nephrotic syndrome, may contribute to slight changes in serum creatinine values. Similarly, cyclosporine starting dose of 5 mg/kg may be too high especially in patient already showing signs of renal compromise, and prompt sudden drop of eGFR could thus be considered as treatment failure. There is also the question of how many patients with unusually rapid loss of renal function entered the study since inclusion criteria stated “a 20 % or greater decline in GFR that was based on at least three measurements over a period of 3 months or longer within the 2 years before the study.” Thus, it is conceivable that a patient that lost eGFR during a 3–6-month period could be included, but it would be atypical for a patient with MN and would require ruling out a superimposed event, e.g., acute renal vein thrombosis, interstitial nephritis, etc. Unfortunately, these concerns cannot be answered by the data provided in the manuscript.
Calcineurin Inhibitors
A number of uncontrolled studies showed that cyclosporine may obtain remission of the nephrotic syndrome in about 60 % of patients with MN [111–119] (Table 5.2). However, in a number of patients, relapse of nephrotic syndrome occurred after cyclosporine was withdrawn. Cattran et al. reported the results of a randomized trial of cyclosporine in MN [120]. Fifty one patients with steroid-resistant MN and nephrotic syndrome were randomized to either 3.5 mg/kg/day of cyclosporine plus low-dose prednisone or placebo plus prednisone for 26 weeks. At the end of the treatment period, 75 % of the treated patients versus 22 % of the placebo patients had a partial or complete remission. After 6 months, relapses of nephrotic syndrome occurred in 43 % of treated and in 40 % of untreated patients. At the end of the study (78 weeks), despite the high relapse rate, the incidence of remissions was still significantly higher in the cyclosporine group than in controls (39 % vs. 13 %). Renal function remained unchanged in both groups during the treatment period but later deteriorated in two patients for each group.
Although short-term, the available studies indicate that cyclosporine may be effective in inducing remission of the nephrotic syndrome. It is possible that a prolonged administration may increase the chances of response to cyclosporine. A German multicenter study reported, showing that in patients with MN, the mean time to response to cyclosporine was in the range of 8 months [112]. It is important to emphasize that although reduction of proteinuria usually occurs within a few weeks, the majority of complete remission occurred after more than 6 months of treatment. On the other hand, if after 3–4 months of cyclosporine therapy at adequate doses, proteinuria is not significantly reduced, it is unlikely that the therapy will be effective. The optimal dose and duration of treatment remain a controversial issue. It should be noted, however, that good results have been obtained with doses of 2–3 mg/kg/day and that the addition of small doses of prednisone may improve the probability of response [118]. Unfortunately, most responders develop relapses of the nephrotic syndrome after the end of cyclosporine administration. However, a number of patients may be maintained in remission if cyclosporine is tapered off very gradually [121]. It remains still unclear whether the use of cyclosporine in MN may have long-term benefits. Repeat biopsy examinations performed in patients responsive to cyclosporine revealed the persistence of immunoglobulin and complement deposits, suggesting that the disease process was not halted by treatment [80].
The main problem with cyclosporine is the risk of nephrotoxicity, which may render the patient more susceptible to the risk of progressive renal failure. This risk is dose-dependent and is particularly increased in patients with elevated plasma creatinine levels and tubulointerstitial lesions at renal biopsy [122]. Moreover, the higher the increase in plasma creatinine levels during cyclosporine treatment, the higher the risk of irreversible nephrotoxicity [122]. Thus, patients with a creatinine clearance lower than 60 ml/min and/or those with severe interstitial fibrosis and tubular atrophy should not be treated with cyclosporine, and caution is recommended in patients with subnormal levels of creatinine clearance and/or moderate tubulointerstitial lesions. In addition, whenever plasma creatinine rises by more than 30 % over the baseline, cyclosporine should be reduced or stopped for at least 1 month in case of further creatinine increase; cyclosporine may be reintroduced only if plasma creatinine returns to normal or to values no higher than 10 % over baseline. In MN the starting dosage of cyclosporine microemulsion should not exceed 4 mg/kg/day. If nephrotic syndrome goes into remission, the doses may be reduced slowly (0.5 mg/kg/day every month) to a maintenance dosage of 2.5–3.5 mg/kg/day. After 1–2 years a cautious trial of stopping cyclosporine may be attempted, by tapering the doses very gradually. If the indications and contraindications are respected, the nephrotoxic risk of cyclosporine treatment appears to be low [121]. Prolonged low-dose cyclosporine (~1.5 mg/kg/day) could be considered for long-term maintenance of patients with preserved renal function who achieve a complete remission or partial remission but that relapse once cyclosporine is discontinued or the blood trough levels are maintained below 100 ng/ml. With such a strategy, there is a little risk of nephrotoxicity [118, 123].
Tacrolimus has also been used. In a pilot study, 21 patients received low doses of oral steroids and tacrolimus with the addition of mycophenolate mofetil in nine patients with proteinuria >1 g/day after 3 months. At the end of the treatment, eight patients (38.0 %) had complete remission and seven (33.3 %) partial remission, but 11 (73 %) responders relapsed [124]. In a randomized trial, 25 patients received low-dose tacrolimus (0.05 mg/kg/d) over 12 months with a 6-month taper, whereas 23 patients were assigned to the control group [125]. The probability of remission in the treatment group was 94 % after 18 months versus only 35 % in the control group. However, nephrotic syndrome reappeared in almost half of the patients who were in remission by the 18th month after tacrolimus withdrawal.
In summary, calcineurin inhibitors represent a promising and perhaps useful therapeutic option for patients with MN but their administration should be prolonged since relapses are frequent after treatment cessation.
Adrenocorticotrophic Hormone
Another approach to the treatment of MN consists in the prolonged administration of synthetic adrenocorticotrophic hormone (ACTH). Berg et al. treated nine patients with an 8-week course of i.m. synthetic ACTH given three times a weeks and gave ACTH twice a week for 1 year to five patients with resistant MN and severe nephrotic syndrome [126]. All patients obtained a median reduction of 80 % in the urinary protein excretion. However, after withdrawal of ACTH, proteinuria relapsed in the patients treated for 8 weeks, whereas it did not relapse in any of the five patients treated for 1 year, during 18 months of observation following the end of treatment.
On the basis of these promising results, a few small-sized studies have been performed. In a pilot study seven patients with MN, nephrotic syndrome, and normal renal function received synthetic ACTH 1 mg twice weekly for 1 year [127]. Two patients had to stop treatment due to allergy and malaise, respectively. All the five patients who completed the ACTH course obtained a complete remission of nephrotic syndrome, which persisted up to 26 months after the end of the ACTH course. An exploratory multicenter randomized trial compared the efficacy and safety of a 6-month treatment with methylprednisolone alternated with a cytotoxic drug, versus 1-year synthetic ACTH treatment at a dose of 1 mg twice a week in 32 patients with biopsy-proven MN and nephrotic syndrome [128]. Complete or partial remission as a first event was attained by 93 % of the 16 patients assigned to the combined therapy and by 87 % of the 16 patients assigned to ACTH. At the last observation, 75 % of patients given the cyclical therapy were in remission (four complete and eight partial) compared with 87 % of patients treated with ACTH (eight complete and six partial). Median proteinuria significantly decreased from the basal value in both groups without any difference between the two groups. In the group receiving the cyclical therapy, two patients did not complete treatment because of severe leukopenia. One patient in the group assigned to ACTH had worsening renal function and had to undergo regular dialysis 18 months after randomization. A patient did not tolerate ACTH because of dizziness, and another patient stopped therapy because of lack of response. Two patients from each group developed glucose intolerance, which reversed after completion of treatment.
In a trial of Arnadottir et al. reported in form of abstract, 30 patients with idiopathic MN and nephrotic syndrome were given ACE inhibitors and statins and were randomized to add synthetic ACTH subcutaneously twice a week for 9 months or to continue with symptomatic therapy [129]. At a follow-up of 21 months, 11 of 15 patients given ACTH were in complete remission and 3 in PR, while only 1 control reached complete remission and another one entered PR. Serum creatinine decreased significantly in the ACTH group and increased significantly in the group treated with ACE inhibitors and statins alone.
All these studies were conducted using a synthetic version of ACTH (Synacthen®; not available in the USA), but a retrospective case series of patients with nephrotic syndrome using a natural, highly purified ACTH gel formulation (H.P. Acthar Gel®), currently approved in the USA for remission of proteinuria in the nephrotic syndrome, reported similar encouraging results [130]. Bomback and colleagues retrospectively reviewed data on 21 patients identified as receiving treatment with Acthar for resistant idiopathic nephrotic syndrome [130]. Of these, 11 patients were identified as having MN that did not respond to previous treatments. The most common treatment regimen used was Acthar Gel® 80 units (U) subcutaneous twice weekly for 6 months. Dosing intervals varied from 2 to 3 times weekly. Most patients were treated for a minimum of 6 months, with the longest treatment period being 14 months. Nine of the eleven patients with MN achieved a complete or partial remission. No severe infections were reported in the entire cohort. A multicenter, randomized, double-blind, 4-arm, placebo-controlled, comparing Acthar® Gel with placebo in treatment-resistant patients with MN and nephrotic syndrome is currently underway (ClinicalTrials.gov Identifier: NCT01386554).
The exact mechanism by which ACTH mediates its effects in proteinuria is not completely understood, but is likely independent of its induction of cortisol production, as its production remains low and there is concomitant evidence that steroids alone do not affect the outcome of the disease [90]. Ponticelli et al. hypothesized that by modifying apolipoprotein metabolism, ACTH might restore glomerular expression of apolipoprotein J (also called clusterin) which is inappropriate in patients with MN [128, 131]. Experimental studies showed that clusterin competes with the terminal components of complement, C5b-9, for the same receptor in podocytes, namely, megalin, which has been identified as the target of the C5b-9 injury in experimental models of MN [132]. Therefore, the presence of clusterin in glomeruli would represent a limitation to the lesions caused by complement, whereas deficient expression could enhance these lesions [128]. More recently, it has been suggested that ACTH may mediate its effects via the α[alpha]-melanocyte-stimulating hormone (α[alpha]-MSH). ACTH is derived from the pro-opiomelanocortin (POMC) precursor. POMC is proteolytically cleaved by endopeptidases to yield various polypeptide fragments with varying physiological activity such as ACTH and α[alpha]-MSH. To date, five forms of melano-corticotropic receptor (MCR) have been cloned, each with different tissue distributions, affinities, and physiological roles. MCR1 is located on various cells, including B cells, T cells, antigen-presenting cells, and human podocytes. In a recent study, treatment with ACTH, α[alpha]-MSH, or MS05, a specific MCR1 agonist, showed similar but significant reduction in proteinuria in rats with passive Heymann nephritis [133]. These results suggest that ACTH mediates its effects via α[alpha]-MSH interaction on MCR1 on podocytes and may explain why patients who are resistant to previous immunosuppressive therapies respond to ACTH. Acthar®, which is obtained from the processing of porcine pituitary, contains a highly purified form of ACTH with smaller amounts of other POMC peptides and ACTH fragments/analogues. Acthar is known to have activity at all five MCRs and all MCRs are known to be activated by more than one melanocortin ligand [134]. The steroidogenesis associated with Acthar and ACTH is mediated by activation of the melanocortin 2 receptor expressed in the adrenal cortex and other tissues. The functional roles of all the peptides in Acthar, however, have not been fully elucidated.
ACTH may also work by modulating autoantibody production. In a pilot study, 14 patients with MN treated with Acthar® Gel for 6 months were evaluated for its effects on anti-PLA2R levels [135]. Twelve patients (86 %) were anti-PLA2R positive at baseline. All 12 patients experienced a reduction in anti-PLA2R (17–100 %) by 6 months with disappearance of anti-PLA2R in five cases. There were five partial remissions at 6 months: three in patients who cleared their anti-PLA2R and two in those lacking baseline anti-PLA2R. Another patient later achieved a 96 % reduction in anti-PLA2R without further treatment while two others, after receiving rituximab for perceived failure of Acthar® Gel, fully cleared anti-PLA2R. With long-term (12–18 months) follow-up of those with undetectable anti-PLA2R at baseline or after Acthar® Gel ± rituximab, there were four complete and two partial remissions, one nonresponder, and a final patient who achieved partial remission but relapsed coincident with a return of anti-PLA2R. Two other patients also showed an increase in anti-PLA2R after discontinuing treatment with Acthar® Gel. Thus, measurement of anti-PLA2R provides useful information relating immunological and clinical disease activity in MN patients treated with new agents such as Acthar® Gel. This study suggests that Acthar® Gel may work in part by suppressing autoantibody production although the duration and degree of this response need further study [14–16].
Intravenous Immunoglobulins
The mechanism of therapeutic effects of intravenous immunoglobulin (IVIg) in MN is not well established. It has been speculated that IVIg afford its immunomodulatory effects through several possible Fc- and F(ab)-mediated mechanisms [136, 137]. There is also evidence that IVIg interferes with complement-mediated immune damage by binding to C3b and C4b. Such an effect would reduce complement-mediated glomerular injury. This mechanism may be involved in MN as suggested by a study in passive Heymann nephritis in which treatment with systemic immunoglobulin obtained a decrease in proteinuria, associated with a decreased glomerular deposition of C3c and C5b-9, without changes in the amount, size, or distribution of the subepithelial immune complexes [138].
Kida et al. first reported the results of two regimens of IVIg therapy in patients with MN [139]. Four patients received a single bolus of 0.5 g/kg repeated for 6 days, and 19 patients were given a single dose of 1 g/kg. Only one of the four patients treated with the first regimen obtained a remission, while 14 out of the 19 patients given 1 g/kg developed remission within 1 year. Palla et al. treated nine patients with MN (five with normal and four with moderate renal insufficiency) with pulse doses of IgG (0.4 g/kg body weight) for 3 consecutive days, repeated three times at 21-day intervals for 10 months [140]. Complete or partial remission of proteinuria was obtained in all of the five patients with normal renal function and in three of four patients with mild renal insufficiency. Interestingly, a second renal biopsy in five responders showed the disappearance of glomerular deposits of IgG and C3 at immunofluorescence, a regression from stage II to stage I in two patients and a complete recovery of the glomerular lesions in three patients. No information about the long-term outcome of these patients was provided. Another retrospective study compared the outcome of 30 patients treated with IVIg, plus corticosteroids and/or alkylating agents in five patients, with the outcome of 56 control patients who received either no treatment (30 %) or treatment with corticosteroids alone or in combination with alkylating agents [141]. The IVIg regimen consisted of 1–3 courses of 5–10 g/day for 6 consecutive days. At 6 months, patients treated with IVIg obtained a statistically significant higher rate of complete remission (57 % vs. 10 %). However, the difference disappeared later. At 12, 24, or 60 months of follow-up, the rate of remission was similar between the two groups. Moreover, the benefits of IVIg were limited to a subgroup of patients with a homogenous (synchronous) pattern of immune deposits.
Although usually well tolerated, treatment with IVIg has potential complications such as acute renal failure, vascular thrombosis, contamination with infective agents, and respiratory distress. Moreover, the cost of intravenous immunoglobulin is elevated.
Mycophenolate Mofetil
The effectiveness of mycophenolate mofetil (MMF) in some animal models has encouraged its evaluation in the treatment of human glomerular disease, including MN. Penny et al. showed that MMF can prevent the induction of active Heymann nephritis in rats treated at the beginning of the disease course, while it did not have a beneficial effect when given later in the course of the disease [142]. In another study, rats given MMF before the induction of active Heymann nephritis developed less proteinuria than untreated rats and showed an attenuated antibody response persisting after cessation of treatment [143].
Some authors have evaluated in small uncontrolled studies the efficacy of MMF in human MN. Obviously, in contrast with the experimental model, human clinical studies examined the response to MMF among patients with established MN. In a study by Miller et al., 16 patients with resistant MN were treated, for a mean of 8 months, with MMF (0.5–2.0 g/day), plus prednisolone in 5 of them [144]. At 6 months, six patients experienced a halving of proteinuria, but only two patients developed a partial remission, and none obtained a complete remission of the nephrotic syndrome. There were not significant changes in the serum creatinine during the study. Eighteen percent of treated patients developed relevant side effects. In another retrospective study, 17 patients received MMF for 6–25 months, at a dosage ranging between 0.5 and 1.5 g twice a day [145]. At the end of treatment, mean proteinuria significantly decreased in all patients. Among the 15 nephrotic patients, 2 achieved a complete remission and 8 a partial remission. In addition, 14 of 15 patients with either corticosteroid or cyclosporine dependency were able to successfully withdraw either agent. However, MMF dependency developed in four patients and three patients had to stop therapy because of severe side effects. However, the efficacy of MMF treatment was difficult to evaluate, since only three patients received MMF monotherapy, whereas most patients were also given variable doses of steroids, cyclosporine, or cytotoxic agents. In another small study, eight nephrotic patients with stage III–IV MN were treated with 2 g/day of MMF for 9 months [146]. After 3 and 9 months of treatment with MMF, proteinuria significantly decreased from the basal value of 4.4 g/day to 2.0, and 1.9 g/day, respectively. Renal function did not change significantly.
Branten et al. compared treatment with IV methylprednisolone 1 g for 3 consecutive days at the beginning of months 1, 3, and 5 followed by alternate-day prednisone, 0.5 mg/k/48 h, for 6 months plus MMF 2 g/day for 12 months (32 patients) or cyclophosphamide 1.5 mg/kg/day for 12 months (32 historical controls) [147]. There were no significant differences in remission of proteinuria at 12 months or in adverse events between the two groups. However, 75 % of the patients treated with MMF relapsed within 2 years after the end of treatment. Similarly, Senthil Nayagam et al. reported that a 6-month course of combined corticosteroids with MMF is as effective as alternating monthly cycles of steroids and cyclophosphamide for 6 months for primary treatment of MN in the short term [148]. On the other hand, Dussol et al. randomized 36 patients with MN and nephrotic syndrome to receive conservative therapy plus MMF (2 g/d) (n = 19) or conservative therapy alone (n = 17) for 12 months and found that the probability of complete or partial remission did not differ between the two groups [149].
In summary, looking at these studies, the impression is that in contrast with the results obtained in the experimental model, treatment with MMF in clinical MN had moderate efficacy, but better results might be obtained by combining MMF with steroids.
Rituximab
In MN, experimental data suggest that B cells are involved in the pathogenesis of the disease [150]. To date, the best proven therapy for patients with MN consists of the combined use of corticosteroids and cyclophosphamide. The mechanism of action of cyclophosphamide includes suppression of various stages of the B cell cycle including B cell activation, proliferation, and differentiation and inhibition of immunoglobulin secretion, supporting the hypothesis that B cell abnormalities are involved in the pathogenesis of MN [151, 152]. Given the key role of IgG antibodies in IMN [2], it is reasonable to postulate that suppression of antibody production that targets glomerular antigens by depleting B cells may improve or even resolve the glomerular pathology. This is the theory underlying the application of B cell targeting with rituximab in MN. Rituximab is a chimeric monoclonal antibody which contains a human Fc IgG1 region and a murine variable region specific for the CD20 B cell antigen. This agent acts through a ligation with the membrane receptor CD 20 of B cells and inhibits their activation, proliferation, differentiation, and immunoglobulin secretion. In contrast to cyclophosphamide that has striking but nonselective effects on B cell function, rituximab offers a more targeted approach to B cell depletion.
Remuzzi et al. studied eight consecutive patients with MN treated with full-dose ACE inhibition, who had persistent nephrotic syndrome for at least 6 months [153]. All patients received 4 weekly infusions of 375 mg/m2 of rituximab. Treatment obtained a significant decrease of urinary protein excretion from 8.6 to 3.8 g/24 h. Two patients achieved a decrease of proteinuria to 1 g/24 h or less and 3 had a decrease to 3.5 g/24 h or less. None of the remaining patients had any worsening of proteinuria. CD20+ lymphocytes decreased to undetectable levels by month 1 after rituximab infusion and remained below normal ranges up to the end of study. Of note, after the completion of the study, one patient had progressive recovery of circulating CD20 lymphocytes to normal ranges that was paralleled by progressively increasing proteinuria to pretreatment values, opening the question if rise in CD20 lymphocytes is inevitably associated by an increase in proteinuria in all patients. In a subsequent paper, Ruggenenti et al. reported that rituximab proved to be ineffective in patients with severe tubulointerstitial lesions at renal biopsy [154].
Two subsequent studies involving a total of 35 patients (about half of them refractory to prior therapy) treated with rituximab 1G IV 2 weeks apart or 375 mg/m2 for 4 weeks showed 50 % complete or partial remission of proteinuria at 1 year and 80 % at 2 years [155, 156]. The response in proteinuria was gradual and sustained and there was no difference in the effectiveness at 1 year between the two different dosing regimens. Total B-cell counts started to recover at 3 months, which is faster than in patients with ANCA-associated vasculitis, rheumatoid arthritis, or non-Hodgkin’s lymphoma suggesting that heavy proteinuria resulted in decreased levels in rituximab, though there was no correlation between rituximab levels, degree of proteinuria, or response to drug. A B-cell-titrated protocol using a single dose of rituximab 1 g has shown to be similarly effective as the 4-dose protocol but at a lower cost [157]. A beneficial effect was also reported in a matched-cohort study compared 2-year outcomes of 11 consecutive patients with primary MN who received rituximab as second-line therapy for persisting nephrotic syndrome or relapsing disease [158]. These encouraging results have been further supported by recent results in the largest study to date which included 100 consecutive patients with nephrotic syndrome treated with rituximab [159]. During a median follow-up of 29 after administration of rituximab, 65 patients achieved complete or partial remission of proteinuria. Remission rates were similar between patients with or without previous immunosuppressive therapy. No serious adverse events were attributed to rituximab [159]. Rituximab may also allow successful withdrawal in calcineurin-inhibitor-dependent patients [160]. These results suggest that rituximab is effective in inducing remission of proteinuria in a large number of patients with MN, either as initial treatment or for patients refractory to previous therapeutic attempts. The short-term side-effect profile and compliance issues of this selective therapy seems much more favorable than the currently used immunosuppressive regimens, although some concerns about the long-term effects of rare and fatal complications such as progressive multifocal leukoencephalitis potentially related to B cell depletion therapy have been reported.
Monitoring anti-PLA2R levels may be also helpful to monitor response to therapy in patients treated with rituximab. Hoxha et al. treated five patients with MN with rituximab; in two of them disappearance of anti-PLA2R from circulation anticipated a complete or partial remission of proteinuria [161]. Patients who failed to clear anti-PLA2R from circulation did not achieve remission of proteinuria. Similar results have been reported in a study involving 35 patients with primary MN treated with 2–4 doses of rituximab [17]. Pretreatment samples contained antibodies against anti-PLA2R in 25 of 35 (71 %) patients. Autoantibodies declined or disappeared in 17 of 25 (68 %) patients within 12 months after rituximab. Those who demonstrated this immunologic response fared better clinically with 59 % and 88 % of the patient attaining complete or partial remission of proteinuria at the end of 12 and 24 months, respectively. This compared with 0 % and 33 % among those patients with persistent anti-PLA2R levels. Changes in antibody levels preceded changes in proteinuria. One subject who relapsed during follow-up had a concomitant return of anti-PLA2R [17] (Fig. 5.4). The relationship between reduction in anti-PLA2R levels and response to rituximab points toward a direct effect on the pathophysiology of the disease process and emphasizes the future of more specific targeting therapy in MN.