Biliary excretion is the only mechanism for copper elimination under physiological conditions, and it increases with increasing size of the hepatic copper pool. Trafficking of copper in and through the hepatocytes involves several transport proteins: Copper transporter 1 (Ctr1) responsible for the copper uptake at the hepatocyte plasma membrane; metallothioneins (MT), intracellular proteins capable of binding metalions, including copper; and metallochaperones that mediate the delivery of copper to specific proteins, and they are responsible for transfer of copper from metallothionein to the site of synthesis of copper containing proteins. Trafficking of copper is essential for further binding with ATP7B protein .
The WD protein, ATP7B has got several functions that finally allow effective copper excretion from the hepatocyte and binding with ceruloplasmin which protects from potential copper toxicity. Copper transport into the trans-Golgi is initiated by ATP binding to the “nucleotide binding domain” of ATP7B. ATP hydrolysis and subsequent dephosphorylation allows the movement of copper associated with two binding sites on one of the transmembrane domains into the lumen of the trans-Golgi. Entering trans-Golgi is essential for the further transport of copper—it is incorporated into apo-ceruloplasmin and other apoproteins and then excreted to sinusoids or after saturation of six copper-binding sites at the N-terminal cytoplasmic tail of ATP7B and phosphorylation of serine residues it migrates in vesicles to the biliary canaliculus [1] .
Mutations of the WD gene coding ATP7B protein impair its function and lead to accumulation of copper in the hepatocyte and in other organs. There are more than 500 mutations in the WD gene identified which affect the catalytic and export functions of the protein (according to the database: http://www.wilsondisease.med.ualberta.ca/database.asp [3]). The most common are missense mutations. The functional consequences of most mutations are not described. Usually two different mutations can be found on two different alleles of a gene (heterozygous composition). The most common mutation in Caucasian patients is H1069Q in the nucleotide-binding domain. This mutant binds ATP in an incorrect orientation with a reduced affinity, causing instability due to temperature-dependent unfolding and retention of ATP7BH1069Q within the endoplasmic reticulum [4]. For better understanding the role of different mutation of functionality of ATP7B, a yeast strain that lacks its endogenous copper transporter was used to segregate ATP7B mutants into severe and mild categories based on their ability to restore growth. Studies in mammalian cells have shown decreased protein levels and mislocalization for several mutants, and copper transport has been studied in vesicles derived from cell lines with various ATP7B mutations [5]. The functional studies can help to understand why some mutations are pathogenic and lead to copper accumulation, but do not yet explain the phenotypic variability of WD. Liver disease seems to result from a direct copper accumulation in hepatocytes, and consequently mitochondrial damage and disturbance of lipid oxidation. Hepatic steatosis is an early pathologic feature of the disease and may be explained by this mechanism. Injury of other tissues seems to be a consequence of copper accumulation outside the liver and usually appears at later age. Once capacity of the liver to store copper is exhausted, copper is released into the circulation. It can be taken by different organs but the central nervous system is most vulnerable [6] .
Specific pathways that allow the intracellular trafficking and compartmentalization of copper within the hepatocyte showing the role of ATPase7B are shown on Fig. 65.1.
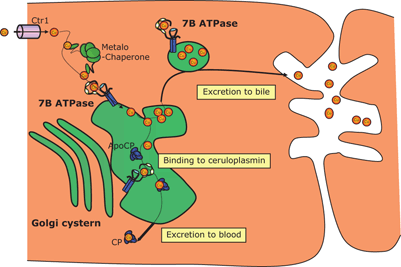
Fig. 65.1
Copper transport within a hepatocyte involving ATPase7B protein
Liver Histology and Ultrastructural Changes
Histological abnormalities observed in liver biopsy are not specific for WD and cannot be regarded as a valuable diagnostic tool. In the early phase, microvesicular and macrovesicular fatty deposition can be observed (Fig. 65.2), with glycogen-containing vacuoles in the nuclei of periportal hepatocytes. Liver disease may then progress to portal fibrosis and inflammation. It should be noticed that histological changes in WD may resemble those observed in autoimmune hepatitis (AIH) with interportal fibrous bridging or cirrhosis.
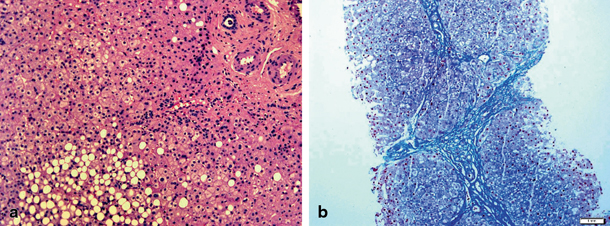
Fig. 65.2
Histopathological findings on liver biopsy from patients with Wilson’s disease: a steatosis (HE staining) and b advanced fibrosis (Azan staining). (Courtesy of Dr Joanna Kuszyk, Department of Pathology of the Children’s Memorial Health Institute, Warsaw)
Utrastructural changes are also not regarded to have a significant diagnostic value— one of the typical features are pleomorphic mitochondria with increased matrix density and widening of intercristal spaces.
Copper staining can be used for diagnostic purposes—using rhodanine or rubeanic acid staining. The absence of histochemically demonstrable copper does not exclude a diagnosis of WD. Liver copper content is used as one of major diagnostic criteria—therefore in suspected WD, a piece of the liver biopsy should be placed in a dry plastic copper-free container for subsequent analysis by atomic absorption analysis.
Clinical Symptoms
WD may present with liver, neurological, or psychiatric symptoms. It seems that liver involvement can be observed in most of the neurological presentations but it is not always looked for. Due to high index of suspicion in many countries WD is diagnosed very early in the presymptomatic phase. WD may present at any age from early childhood (with raised transaminases, measured for some unrelated reason) to the eighth decade (often with surprisingly mild neurological features). Still, the clear liver symptoms (like hepatomegaly, clotting disturbances) have not been observed before the age of 3 years.
Hepatic Symptoms
There is a wide spectrum of hepatic involvement from asymptomatic to hepatomegaly, fatty liver disease, hepatitis, jaundice, cirrhosis, and liver failure at the end . As indicated earlier, the liver damage and liver symptoms are not characteristic and making diagnosis required biochemical and/or molecular tests. AIH can be easily misdiagnosed as specific for this condition autoantibodies are commonly found in WD. Therefore final diagnosis of AIH requires exclusion of WD.
Acute liver failure is a severe presentation of WD and requires fast diagnosis which may be difficult to obtain. It is usually found in a previously healthy child who suddenly presents with jaundice, hepatomegaly, rapidly progressing coagulopathy, and, in some cases, encephalopathy. The case history of a child may reveal important clinical problems in the past: episodes of jaundice, hemolytic anemia, or increased transaminases. Even if acute liver failure is not a chronic condition, liver biopsies taken at transplantation or postmortem show cirrhosis which indicates that it is acute on chronic liver disease. Acute liver failure is defined according to the Pediatric Acute Liver Failure (PALF) study group—in a child with biochemical evidence of acute liver injury, international normalized ratio (INR) exceeds/equals 1.0, in the presence of clinical hepatic encephalopathy (HE), or exceeds/equals 2.0 regardless of encephalopathy (HE). WD makes a significant proportion of ALF etiologies—in 703 ALF patients in the PALF registry, 23 had a diagnosis of WD. However, among 329 patients without a final diagnosis, WD had been tested for in only 81 % so there may be some underdiagnosis [7]. WD comprised 4 % of the King’s College Hospital pediatric ALF series [8].
Some clinical and lab features may indicate WD in acute liver failure like KF ring, family history of WD, neurologic features of WD, jaundice and relatively moderately increased transaminases (100–500 IU/l) and alkaline phosphatase (< 600 IU/l), hemolysis, and high bilirubin (> 300 μmol/l) [9] .
Presence of encephalopathy is a bad prognostic feature [10].
Chronic liver disease is more common among Wilsonian patients. Again, AIH may be falsely diagnosed because of the presence of low-titre autoantibodies. The differential diagnosis requires also testing for viral hepatitis and alpha-1-antitrypsin deficiency.
Acute hepatitis is not commonly observed, and it can be rather looked for in the past history of the patient with WD who could have had an episode of jaundice and malaise from which he recovered .
Neurological Symptoms
Neurological symptoms are extremely rare in children with WD as they usually develop in the third decade of life and are the presenting symptoms of WD in about half of all patients . Some initial symptoms can be difficult to be diagnosed like difficulty with speech. Otherwise the neurological symptoms seem to be typical and can be described as:
1.
A dystonic syndrome with dystonic postures and choreoathetosis
2.
An ataxic syndrome presenting as postural and intentional tremor and ataxia of the limbs
3.
A parkinsonian syndrome with hypokinesia, rigidity, and resting tremor
To improve diagnostic approach and evaluate therapy, Czlonkowska and coworkers developed a scoring system for neurologic symptoms in adult patients with WD based on the Eurowilson project [11, 12]. The limited data on neurological presentations in children does not allow describing the frequency of neurological symptoms at this age. However, dysarthria, salivation, gait disturbances, and postural tremor should be looked for. The presence of Kayser–Fleischer ring is usually associated with neurological involvement .
Other Symptoms
Psychiatric symptoms are described to be the predominant presentation of WD recognized in adults in later age but usually are associated with other clinical symptoms . The most common seems to be depression which can lead to attempted suicide (also from author’s experience in children). Psychiatric problems are not easy to detect as some of them can be attributed to the teenage such as mood change, aggressiveness, and irritability. These symptoms may also be a reason for noncompliance with therapy once WD is diagnosed.
Kayser–Fleischer ring is very typical for WD and should be always looked for with a slit lamp—it is a gold or gray-brown opacity in the peripheral cornea which represents a deposit of copper and sulfur-rich granules in Descemet’s membrane. Although very characteristic, Kayser–Fleischer ring is uncommon in children (observed in less than 5 % of children with WD) .
Hemolysis with negative Coombs test is another typical feature of WD even if not commonly described in children with WD—usually it associated with fulminant liver failure.
Dermatological findings are also described in children with WD such as xerosis, keratosis piliaris, spider angioma, papulopustular lesions, and hyperpigmentation [13].
Fanconi syndrome (renal tubular abnormality presenting as glycosuria, aminoaciduria, renal tubular acidosis, impaired phosphate reabsorption) can be observed in the course of WD. Proteinuria may also be detected pointing to glomerular damage in the course of WD but it is usually associated with penicillamine therapy.
Skeletal manifestations appear rarely and include arthritis, rickets, or osteoporosis.
Asymptomatic WD
Asymptomatic liver disease seems to be the most common presentation due to increasing awareness of WD manifestation with slightly increased transaminases . Abnormalities at physical examination like hepatomegaly and splenomegaly and abnormal liver tests raise a suspicion of WD. In some countries, liver tests are checked at many occasions and any abnormalities lead to further differential diagnosis of liver problems [14]. Siblings of Wilsonian patients should be also diagnosed as early as possible usually in the preclinical phase.
Diagnostic Approach
As WD can mimic any kind of liver disease in childhood other than neonatal cholestasis , the diagnosis rests upon lab findings (Table 65.1) [15]. Ensure that the lab you are relying upon participates in an external quality assurance scheme. Each of the tests has limitations, and particular tests vary in usefulness in different clinical situations.
Test | Comments |
|---|---|
Serum ceruloplasmin | < 20 mg/dl (in > 80 % of WD patients) |
Urinary copper excretion | 24 h copper excretion > 100 µg in 65 % of WD patients |
Urinary copper penicillamine challenge with two dosages of 500 mg 12 h apart and measure urine copper | 24 h copper excretion > 1600 µg in patients with active liver disease |
Serum copper | Serum copper may be low in asymptomatic cases (because ceruloplasmin is low) or high in cases with active liver disease (because free copper is raised) |
Serum “free” copper calculated on the basis that ceruloplasmin contains 0.3 % copper | Free copper > 25 µg/dl |
Liver copper | > 250 µg/g of dry weight liver |
Plasma Ceruloplasmin
Plasma ceruloplasmin (CP) may be measured immunologically or by enzymatic activity; the latter is more accurate in WD, where apoceruloplasmin may contribute to the level measured immunologically . Be aware that CP is an acute phase reactant, so may be elevated into the normal range in WD with active liver inflammation, is produced in the liver so may be reduced in cirrhosis of other causes, may be reduced in WD or aceruloplasminemia heterozygotes, may be low in protein losing enteropathy, and is a glycoprotein so may be reduced in some disorders of glycosylation. Twenty percent of WD patients may present with normal ceruloplasmin levels. Values exceeding 30 mg/dl are rare in WD .
Twenty-Four-Hour Urinary Copper Excretion
Accuracy of 24 h urinary copper estimations depends on the collection, the container, and the lab. Values greater than 40 μg/24 h raise the suspicion of WD; values greater than 100 μg/24 h make it very likely .
In the originally described penicillamine challenge test using a cut-off value of 25 mcmol/24 h (1600 mcg/24 h), the test was abnormal in 15 of 17 Wilsonian patients with active disease and 1 of 58 non-Wilsonian patients. The test was again evaluated by Muller et al. who showed sensitivity to be 76 % and specificity—93 % in the whole cohort of patients. However, and most importantly, the sensitivity was as high as 92 % in symptomatic patients and only 46 % in asymptomatic patients [16]. Others have found a cut-off value five times the upper limit of normal gives good differentiation. The test has not been evaluated in adult neurologically presenting cases .
Serum Copper
Serum copper is largely CP bound, so will be low in mild or presymptomatic disease but sometimes raised (e.g. in acute liver failure) if serum-“free” copper is raised . Calculated free serum copper according to the formula (total copper − 0.3 % ceruloplasmin) is in practice a disappointingly inaccurate parameter, but the recently described direct measurement of “relatively exchangeable copper” holds more promise [17].
Liver Copper
When a liver biopsy is performed in suspected WD patients, a specimen for measurement of liver copper should always be obtained . Values equal to or higher than 250 µg/gm of dry weight are considered to be typical for WD. In chronic cholestatic conditions, the liver copper content will also be elevated, but this should not be a source of diagnostic confusion. Values less than 250 μg/g may be found in WD cirrhosis, where the centers of large regenerating nodules and tracts of fibrous tissue will both have lower Cu concentrations. Thus, Ferenci et al. assessed the hepatic copper content of 106 patients at the time of diagnosis of WD of whom 19 Wilsonian patients had a liver copper concentration below 250 µg/g dry weight. The sensitivity analysis based on comparison of these 106 patients to 244 other patients without WD showed that the upper limit of diagnosis ( > 250 µg/g dry weight) has a poor sensitivity (82 %) and very good specificity. The low range (50 µg/g dry weight) has a higher sensitivity, but lower specificity as well as a positive predictive value [18].
Liver biopsy is rarely performed in adult neurological patients so the value of liver copper quantification in neurological presentation is not established .
Mutations in the WD ATP7B Gene, Locus 13q14.3 and Genetic Testing Strategy
The WD gene comprises 80,000 base pairs on 21 protein coding exons and a poorly characterized promoter . More than 500 mutations are recorded in the WD Mutation Database [3]. H1069Q (hist1069glu) is the commonest worldwide and by far the commonest WD mutation in Central Europe. By contrast, H1069Q is rare in Asian, Japanese, and Chinese patients, where other mutations predominate. In Western Europe, many different mutations are found and patients are often compound heterozygotes. A total of 116 different mutations, including 32 novel mutations, were identified on 356 alleles from 179 individuals in the coding region or adjacent splice sites of ATP7B in the NHS lab serving the UK [19]. Only 40 of these patients were homozygotes, the rest being compound heterozygotes.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


