Fig. 70.1
Pathogenesis of liver cell injury is multifactorial with several factors influencing liver cell death and regeneration. When the net result is liver cell loss beyond a critical mass required maintain normal body function, liver failure sets in
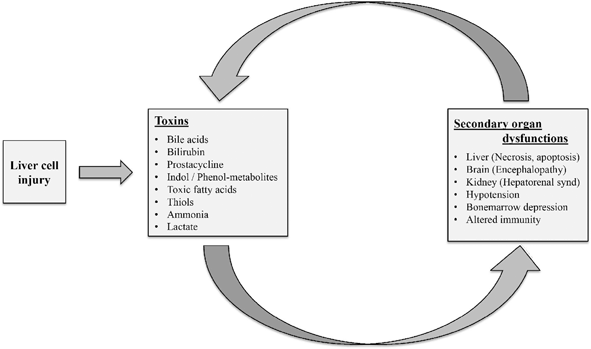
Fig. 70.2
Except for Von Willebrand factor and tissue plasminogen activator, liver is the major site for synthesis of clotting factors. Factors V synthesis is first to be affected and factor VII have the shortest half-lives and are theoretically more sensitive markers than INR of hepatic synthetic function. INR international normalized ratio
Definition
“Fulminant liver failure” was coined by Trey and Davidson 40 years ago to define onset of hepatic encephalopathy (HE) within 8 weeks of appearance of symptoms of liver dysfunction in an otherwise healthy individual with no prior history of liver disease . O’Grady and colleagues categorized ALF as “hyperacute (1 week), acute (1–4 weeks) and subacute failure (5–12 weeks)” depending on the interval between jaundice and onset of encephalopathy, for prognostication purpose [2]. It is difficult to use adult-based definitions in children, as encephalopathy is a late sign and sometimes ALF has in utero onset and so time quantification would be difficult [3]. Acknowledging the fact that diagnosing encephalopathy in children is difficult and often a late and terminal event, Bhaduri and Vergani proposed the definition of ALF as “A rare multisystem disorder in which severe impairment of liver function with or without encephalopathy, occurs in association with hepatocellular necrosis in a patient with no recognized underlying chronic liver disease” [4]. Even this definition does not seemed to be clear as the term “severe impairment of liver function” in the definition did not have any objective quantification values of liver function and so there was a lot of subjective variation in diagnosing ALF .
The pediatric acute liver failure study group (PALFSG) developed a working definition to identify ALF in children without interobserver variation. PALFSG used international normalized ratio (INR) in the background of acute liver disease as an objective measurement to demarcate acute hepatitis and ALF. As per PALFSG, ALF is defined as (i) hepatic-based coagulopathy defined as a prothrombin time (PT) 15 s or INR 1.5 not corrected by vitamin K in the presence of clinical HE or a PT 20 s or INR 2.0 regardless of the presence or absence of clinical HE, [5] (ii) biochemical evidence of acute liver injury, and (iii) no known evidence of chronic liver disease[6]. Asymptomatic preexisting liver disease, which manifests acutely, should be considered as ALF, if they fulfill the diagnostic criteria, as in acute fulminant Wilson’s disease .
Coagulopathy is not only a key criterion in diagnosing pediatric ALF but also acts as a prognostic marker. Due to short half of several liver-based clotting factors, PT/INR functions as a dynamic marker of synthetic inadequacy due to loss of liver cells in ALF. Factors II, VII, IX, and X depends on vitamin K to convert them into active form. Correction of coagulopathy by intravenous vitamin K differentiates between vitamin K deficiency due to decreased absorption from liver synthetic failure. Isolated prolonged APTR is not due to liver disease, as factor VII in extrinsic pathway (Fig. 70.3) has the shortest half-life (4–6 h) of the vitamin K-dependent factors, therefore is the first factor depleted in ALF and invariably affects INR. Coagulopathy secondary to disseminated intravascular coagulation (DIC) precipitated by infection is common in acute liver injury and so it is essential to rule out DIC before making a diagnosis of ALF based on INR .
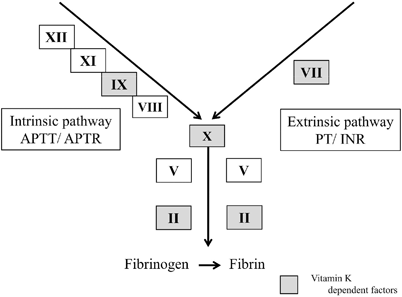
Fig. 70.3
Liver cell injury leads on to release several toxic substances that causes secondary organ dysfunction, this leads on to a vicious cycle of autointoxication. APTT Activated Partial Thromboplastin Time, APTR Activated Partial Thromboplastin Ratio, PT prothrombin time, INR international normalized ratio
Etiology
The etiologies of ALF differ with age groups and geographic location . In Southeast Asia and Latin America, viral hepatitis A and E are the most common cause of ALF in children while in Northern America and Europe etiology remains elusive (indeterminate) in majority of children [7]. Certain disorders, such as neonatal hemochromatosis, are very unique to pediatric population. The exact incidence of ALF in pediatric age group is not known but probably the overall annual incidence of ALF in USA is around 5.5/million population among all ages [8] .
Infection
Hepatitis A and E viral (HAV and HEV) infections are the common cause of ALF in developing countries with poor sanitation and overcrowding, as these viruses are spread by contaminated water and food. The risk of liver failure is 0.1–0.4 % following symptomatic HAV infection and it increases with a preexisting liver disease. Specific diagnosis is established by detecting HAV immunoglobulin (Ig)M antibodies in the blood at presentation. The infection is most often self-limiting with subsequent recovery and only in few it might be severe enough requiring liver transplantation [9]. The risk of developing ALF in adults following HEV infection is 0.6–2.8 %, with higher risk if contracted during pregnancy [10]. Bhatia et al. showed that the case-fatality associated with HEV-induced ALF in pregnancy is similar to that of age-matched general population [11].
The ALF due to hepatitis B virus (HBV) can occur at the time of acute infection, reactivation of chronic HBV infection or seroconversion from a hepatitis Be antigen–positive to a hepatitis Be antibody (HBeAb)-positive state. Infants born to HBeAb-positive mothers are at special risk and could present with ALF around 6 weeks to 9 months [12]. Super infection or coinfection of hepatitis delta virus (HDV) in HBV-infected patients can cause liver failure. Hepatitis C viral (HCV) infection has not been reported as a cause of ALF.
Herpes simplex virus 1 and 2 (HSV) is the predominant cause of viral-induced ALF during first month of life. Neonatal ALF due to HSV carries a high mortality of about 85 % and should be suspected in any neonate with or without vesicular rash who is unwell with dramatically high transaminases and coagulopathy. Treatment with high doses of aciclovir should be initiated in all infants with ALF, while awaiting serology results as the associated liver failure is rapidly fatal. Liver transplantation has a good outcome when considered in a hemodynamically stable neonate with ALF due to HSV [13]. Other members of herpes virus family such as cytomegalovirus, Epstein–Barr virus, and varicella-zoster virus can cause ALF. Rarely, viruses such as Dengue, Lassa, Ebola, Marburg, and Toga and bacteria such as Leptospira and Salmonella are implicated as a cause for ALF.
Drugs and Toxins
In developed countries, drug-induced liver failure is the most common identifiable cause of ALF in adults and children. Drug hepatoxicity could be an idiosyncratic reaction, a dose-dependent response, or a synergistic reaction due of several drugs. It is essential to include details of complementary therapies and herbal medications as some are potential hepatotoxics [14]. Acetaminophen is the most common drug associated with ALF, probably due to the easy availability without the need for a prescription. It is safe when used in recommended doses and toxicity is usually dose dependent. Acetaminophen is detoxified mainly by glucuronidation (40 %), sulfation (20–40 %), and N-hydroxylation (15 %). A small fraction is metabolized via cytochrome P450 to yield N-acetyl-para-benzoquinone-imide (NAPQI), a toxic intermediate compound which irreversibly conjugates with the sulfhydryl group of glutathione and causes hepatocyte necrosis [15]. Genetic polymorphism of cytochrome P450 isoenzymes predisposes affected people to acetaminophen toxicity due to increased NAPQI production. Bound NAPQI forms acetaminophen–protein adducts, which acts as a specific biomarker for chronic acetaminophen toxicity. Hepatotoxicity of acetaminophen is dose dependent and liver injury results either because of acute higher doses ingestion or cumulative over doses taken over a few days. In acute acetaminophen overdose plotting serum levels after 4 h against time on the Rumack nomogram will guide towards potential toxicity. However, it may not be useful in situations of cumulative over doses. In such cases, bound NAPQI forms of acetaminophen–protein adducts in blood would aid the diagnosis. The mechanism of toxicity of anti-tuberculosis drugs, particularly isoniazid is similar to acetaminophen, oxidation via cytochrome P450 pathway results in toxic metabolites.
Most common cause of idiosyncratic drug-induced liver injury (DILI) are due to antibiotics and NSAIDs [14]. The reported incidence of idiosyncratic DILI was around 14 new cases/100,000/year, of which 8 % would progress to ALF [16, 17]. Genetic susceptibility of an individual, unmasking of underlying mitochondrial cytopathies by certain drugs are some of the proposed causes of DILI [18]. Drugs such as diclofenac, mefenamic acid, valproic acid, amiodarone, isoniazid, etc. can unmask mitochondrial cytopathy and cause ALF. Chemotherapy drugs are known to produce veno-occlusive disease leading on to ALF due to endothelial damage.
Councils for International Organizations of Medical Sciences/Roussel Uclaf Causality Assessment Method (CIOMS/RUCAM) scale are helpful in establishing causal relationship between offending drug and liver damage. Using the scoring system, suspected drug could be categorized into “definite or highly probable” (score > 8), “probable” (score 6–8), “possible” (score 3–5), “unlikely” (score 1–2), and “excluded” (score 0) [19]. This scale is helpful in identifying drug-induced hepatotoxicity even in newly marketed drugs and for a previously unreported older drug.
Metabolic Disorders
Metabolic disorders presenting as ALF is common in young children . Galactosemia and tyrosinemia 1 are the most common cause of neonatal liver failure, presenting with hepatomegaly, jaundice, and coagulopathy. Classical galactosemia is an autosomal recessive condition resulting because of mutation in galactose-1-phosphate uridyl transferase gene located on chromosome 9p13. Liver failure in this condition is thought to be due to accumulation on galactitol . Neonates presenting with hepatitis or ALF should be started on a galactose-free formula until galactosemia. Galactose-free diet coupled with supportive management helps the liver to recover, but some may still progress and need a liver transplant. Despite strict galactose-free diet, patients could develop complications like developmental delay, motor disorders and hypergonadotrophic hypogonadism due to endogenous galactose production on a long-term follow-up [20] . Tyrosinemia 1 is an autosomal recessive condition due to defect in the enzyme fumarylacetoacetate hydroxylase. This causes accumulation of intermediate compounds, maleylacetoacetic acid, and fumarylacetoacetic acid, which are then converted to succinylacetone, a toxin that damages the liver and kidneys . Tyrosinemia should be suspected when a coagulopathy and modest rise of transaminases is associated with elevated alpha-fetoprotein levels. Fructosinemia, inborn errors of bile acid synthesis are a few other rare cause of ALF during infantile period.
Medium-chain acyl-coenzyme A dehydrogenases (MCAD) are group of enzymes involved in β-oxidation of 6–12 carbon chain fatty acids in mitochondria . They help in ketone production from fatty acids when hepatic glycogen stores become depleted during prolonged fasting and periods of higher energy demands. Affected children could present with hypoketotic hypoglycemia, recurrent liver failure, precipitated by otherwise minor illness. Unless treated with dextrose supplementation, these episodes may quickly progress to coma and death .
Wilson’s disease, an autosomal recessive disorder could present as ALF. The acute hepatic presentation is usually characterized by the presence of liver failure, Coombs-negative hemolytic anemia and low serum alkaline phosphatase. Diagnosis might be difficult as blood test might show weakly positive autoantibodies and tissue copper estimation might not be possible due to coagulopathy. Demonstration of Kayser–Fleischer rings is diagnostic of Wilson’s disease in a patient who presents with ALF. Wilson’s disease presenting with ALF has high mortality without transplantation .
Mitochondrial disorders are group of spontaneous or inherited disorders of mitochondrial proteins resulting in defective oxidative phosphorylation, fatty acid oxidation, urea cycle, and other mitochondrial pathways [21] . This can affect the function of various cell types, such as neurons, myocytes, etc., where the need for energy requirement is high. Deficiencies of complex I, III and IV, multiple complex deficiencies and mitochondrial DNA (mtDNA) depletion syndrome is associated with liver failure. Diagnosis might be difficult due to particularly in (mtDNA) depletion syndrome where there is tissue specific mitochondrial enzyme deficiency. The infants usually presents with hypotonia, hypoglycemia, feeding difficulties, seizures, and deranged liver function . Liver transplantation could be offered in isolated liver-based mitochondrial disorders, while in multisystemic involvement it should be deferred, hence it is utmost important to perform thorough investigation to rule out neuromuscular involvement [22] .
Gestational Alloimmune Liver Disease
Neonatal hemochromatosis (NH) is the single most common cause of ALF during first month of life, where there is massive iron deposition in liver and extrahepatic tissues, but with sparing of the reticuloendothelial system. The pattern of iron overloading is similar to hereditary hemochromatosis, but NH affects only newborn and so far no specific genetic mutation has been identified. NH was considered to be a primary disease of iron overload of liver leading on to liver failure. Under current concept, iron accumulation in NH is considered to be a secondary phenomenon due to immune-related severe fetal liver injury resulting in impaired regulation of maternofetal iron flux [23]. NH is now referred as gestational alloimmune liver disease (GALD), as maternal antibody is directed towards fetal liver antigen resulting in activation of fetal complement leading to the formation of membrane attack complex (MAC) resulting in hepatocyte loss [24, 25]. This hypothesis is supported by successful prevention of severe disease by antenatal and postnatal treatment with intravenous Ig. GALD presents with jaundice, coagulopathy, moderately elevated alanine aminotransferase, high ferritin, and raised iron saturation levels. High ferritin is seen in other cause of neonatal liver failure and no single biochemical test is diagnostic of NH. The diagnosis can only be confirmed by demonstration of extra hepatic iron deposits sparing the reticuloendothelial system. Labial salivary gland biopsy is safe and effective way of demonstrating this [26]. The disease varies in severity; at one end of spectrum it is associated with fetal death while at the other end spontaneous recovery is reported.
Malignancies
Hemophagocytic lymphohistiocytosis (HLH) is a malignant disorder of hemopoietic system where there is uncontrolled proliferation of activated lymphocytes and macrophages and could present as ALF, particularly during infancy. HLH could be inherited or acquired following infection due to over activation of natural killer cells and of CD8+ T cell lymphocytes, invariably leading to clinical and hematologic alterations. It is associated with defective apoptosis and reduced cytotoxic activity. Familial HLH is an autosomal recessive disease seen mostly in infancy and early childhood. The mutations result in reduced or defective production of cytoplasmic granules such as perforin in cytotoxic cells resulting in paradoxical over activation. Familial HLH is classified into four types based on mutation analysis. Secondary HLH usually occurs after systemic infection or immunodeficiency, which can affect people at any age and may subside spontaneously. HLH presents with fever, cutaneous rash, hepatosplenomegaly, pancytopenia and, in severe cases, with ALF [27]. ALF might be the presenting feature of hematologic malignancies such as leukemia or lymphoma [28]. Usual associated features would be fever, hepatosplenomegaly, high alkaline phosphatase, high lactate dehydrogenase, and abnormalities on peripheral blood film. Bone marrow examination confirms the diagnosis.
Autoimmune Hepatitis
Autoimmune hepatitis (AIH) can present as ALF, most of these children have positive liver–kidney microsomal (LKM) antibody (type 2 AIH). Diagnosis might be difficult, as some may not show specific antibodies at initial presentation. ALF due to AIH without encephalopathy could be benefited by immunosuppression, while ALF along with encephalopathy do not respond to any form of immunosuppression and need urgent liver transplant [29]. In our experience, of the six AIH children presenting with ALF and encephalopathy, four required liver transplantation, one died while awaiting transplantation, and one recovered with steroids [30]. The steroid responder was antinuclear/smooth muscle antibody positive (type 1 AIH) while rest five were LKM positive (Type 2 AIH).
Other Causes
In spite of extensive investigation, the diagnosis could not be found in many children (indeterminate). Indeterminate ALF is probably due to unidentified infectious agent as suggested by presentation with severe hepatitis, liver failure and bone marrow failure mimicking viral-induced disease or presentation with minimal jaundice and centrilobular necrosis on histology suggesting drug induced. There is wide variation in reporting of indeterminate etiology among various centers. This is probably due to incomplete investigations in ALF, which has been highlighted by Narkewicz et al., and labeling them as indeterminate etiology [31]. Hypoxia, bacterial infections, underlying cardiac problem, venous outflow obstruction are few other causes of ALF.
Investigations
As the etiology of ALF is so diverse that it is practically impossible to do all the tests during initial evaluation. A detailed clinical history and thorough general examination could give valuable clue of underlying problem and could help in directing management while awaiting confirmatory results. The first-line investigations should include complete blood count, liver function tests, serum electrolytes , uric acid, lactate, cholesterol/triglyceride, amylase, coagulation studies (INR), and blood glucose. Surveillance blood and urine cultures should be collected prior to starting antibiotics. The clinical presentation along with the results of first-line investigations will guide further specialized tests. Investigations to establish etiological diagnosis are outlined in Table 70.1. Supportive management along with anticipatory management of possible complications associated with liver failure would help in favorable outcome.
Table 70.1
Disease specific investigations in acute liver failure
Infective: |
Serologic/quantitative tests |
Hepatitis A: anti-HAV IgM antibody |
Hepatitis B: HBsAg, HBcAb(IgM), HBcAg |
Hepatitis C: anti-hep C antibody, hep C PCR |
Hepatitis D: anti-hep D antibody |
Hepatitis E: anti-HEV antibody(IgM) |
Human immunodeficiency virus (HIV) |
Herpes simplex virus (neonates) |
Cytomegalovirus, Epstein–Barr virus |
If indicated: measles/varicella/adenovirus/echovirus/leptospirosis/ |
Cultures |
Bacterial cultures: blood, urine, stool, throat swab, sputum |
Skin lesion if present, ascitic fluid if present |
Viral culture of urine and skin lesion if present |
Metabolic: |
Galactosemia: galactose-1-phosphate uridyl transferase |
Tyrosinemia: urinary succinylacetone |
Fructose intolerance: quantitative enzyme assay, q22.3 band mutation in chr 9 |
Mitochondrial disorders: quantitative mitochondrial DNA assay, mutation analysis |
Congenital disorders of glycosylation: transferrin isoelectrophorosis |
MCAD deficiency: plasma acylcarnitine |
Wilson’s disease: serum copper and ceruloplasmin |
24-h urinary copper pre and post penicillamine |
Type 1 and 2 autoimmune hepatitis: |
Immunoglobulins |
Antinuclear antibodies |
Smooth muscle antibody |
Liver cytosol antibodies |
Soluble liver antigen |
Liver–kidney microsomal antibody |
Antineutrophil cytoplasmic antibodies |
Hematological malignancy: |
Bone marrow examination |
Ascitic or cerebrospinal fluid cytospin |
Genetics for HLH |
Drugs and toxins: drug levels in serum/urine |
Budd–Chiari syndrome: ultrasound, echocardiography, computer tomography |
Neonatal hemochromatosis: lip biopsy |
Prognosis
Categorical demarcation between spontaneous liver recovery and irreversible ALF is difficult. In adults with non-acetaminophen-induced ALF, King’s College Hospital criteria (KCHC; Table 70.2) is used for prognostication and the need for liver transplantation. Fulfillment of KCHC is usually associated with death unless transplanted [32]. But in children KCHC does not reliably predict death with a poor positive predictive value of 33 % [33]. Of the several prognostic markers that has been proposed to predict outcomes in ALF in children, INR and factor V concentration remains the best indicators. In children with ALF, INR 4, bilirubin 235 μmol/L, age < 2 years, and WBC > 9 × 109/L are associated with poor outcome without liver transplantation [34]. Bhaduri and Mieli-Vergani have shown that the maximum INR reached during the course of illness was the most sensitive predictor of the outcome, with 73 % of children with an INR less than 4 surviving compared with only 4 of 24 (16.6 %) with an INR greater than 4 [35]. French centers use factor V concentration for prognostication and a value of less than 20 % of normal (Clichy criteria) suggests a poor outcome. New Wilson index proposed by Dhawan et al. based on serum bilirubin, serum albumin, INR, aspartate aminotransferase (AST), and white cell count (WCC) at presentation identified a cutoff score of 11 for death and proved to be 93 % sensitive and 98 % specific, with a positive predictive value of 88 % (Table 70.3) [36]. In acetaminophen overdose, metabolic acidosis with arterial pH less than 7.3, after the second day of overdose in adequately hydrated patients, is associated with 90 % mortality. In acetaminophen overdose, KCHC could be used in children for selecting candidates requiring liver transplantation (Table 70.2).
Table 70.2
King’s College Hospital criteria for liver transplantation
Non-acetaminophen acute liver failure |
INR greater than 6.5 or |
Three of the following five criteria: < div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|




