Differential
The most common mimic of the globules of α1-antitrypsin deficiency is megamitochondria. The location can often be a helpful clue because megamitochondria tend to be randomly distributed, whereas the α1-antitrypsin globules will have a periportal predominance. Special stains can also be helpful, including a phosphotungstic acid hematoxylin (PTAH) for megamitochondria and a PASD for α1-antitrypsin globules. Other rare potential mimics include globular amyloid, fibrin globules of fibrinogen storage disease, and the globules that can be seen with chronic congestive liver disease. These latter globules tend to have a zone 3 distribution and are PASD-negative. Globular amyloid can be highlighted with a Congo red stain or a LECT2 immunostain.
Special Stains
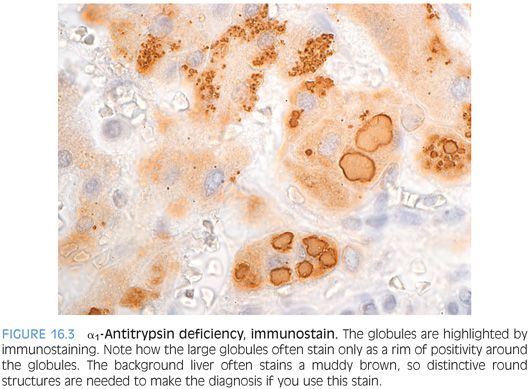
The classic stain for diagnosing α1-antitrypsin deficiency is a periodic acid–Schiff (PAS) stain with diastase treatment (PASD), which highlights the α1-antitrypsin proteins as bright magenta globules (Fig. 16.2, eFigs. 16.2 and 16.3). If the stain is overdigested, however, a false negative can result. The best place to look for the globules is in the periportal hepatocytes. They can be somewhat patchy in early disease, so multiple portal areas should be examined. Interestingly, the globules can also be somewhat patchy in cirrhotic livers, and once again, it is important to check the whole section and to not simply spot-check a few areas. An immunostain for α1-antitrypsin can also be helpful but tends to have high background (Fig. 16.3). Also of note, PASD stains and immunostains can be negative in infants younger than 3 months of age.

WILSON DISEASE
Definition and Mechanism
Wilson disease is caused by mutations in the gene ATPB7, which leads to copper accumulation in affected individuals. The gene ATPB7 encodes an ATPase that both transports copper into the bile for excretion and also helps incorporate copper into ceruloplasmin. Because bile is the main route for copper excretion, the lack of ATPB7 expression leads to a marked reduction in the amount of copper in the bile, with retention of copper in the liver. Mutations also lead to increased copper that is not bound to ceruloplasmin in the blood stream. This free copper can then precipitate in different organs such as the brain, eyes, and kidneys.
Overall, 90% of individuals with Wilson disease will have a mutation detected with full sequencing of the gene. The remaining 10% presumably have mutations in other genes that affect the same metabolic pathway. Wilson disease is autosomal recessively inherited, with an estimated carrier frequency of 1 in 100 individuals. Because of the relatively high carrier frequency, there can be families with successive generations that are affected by Wilson disease, despite the recessive inheritance pattern.1 Heterozygotes may have mild biochemical abnormalities in copper metabolism, but they are clinically unaffected in most cases. More than 500 mutations have been reported in the ATPB7 gene, of which 379 are thought to be disease-causing. Genetic testing for clinical diagnosis is difficult because of the large number of mutations as well as the observation that not all mutations are disease-causing.2 However, once a mutation is identified in an individual with copper overload, focused genetic testing can be very helpful for screening the extended family. There are hotspot mutations that tend to correlate with ethnicity, which can also help focus the genetic screens for probands. For example, H1096Q is present in about 50% of Caucasians with Wilson disease but is rare in Chinese, who tend to have the R778L mutation.
Clinical Findings
Most individuals present between the ages of 5 and 35 years, but individuals can present as young as 3 years of age and as late as 80 years of age.3 The clinical presentations vary considerably but tend to fall into a broad categories of either neurologic symptoms and/or liver disease. Liver disease often is the main finding in younger individuals, with neurologic disease increasing in frequency with increasing age. In terms of liver disease, presentation patterns include acute hepatitis, chronic unexplained elevation in liver enzymes, and cryptogenic cirrhosis.
The main sites of copper accumulation are the liver and the basal ganglia, but the eyes, kidneys, and heart can also be affected. Eye disease manifests as copper deposits in the cornea, which are called Kayser-Fleischer rings. Kayser-Fleischer rings are seen in about two-thirds of individuals with Wilson disease overall but are present in almost all individuals with neurologic or psychiatric symptoms and about 50% of those with liver predominant disease. Although Kayser-Fleischer rings are often considered pathognomonic, there are a variety of mimics that can lead to a false-positive eye exam.4 Important laboratory findings include a low serum ceruloplasmin and elevated 24-hour urine copper. Because ceruloplasmin is an acute phase reactant, levels can be normal or even elevated in individuals with Wilson disease who have significant active ongoing liver inflammation. Estrogens also increase ceruloplasmin levels, and they can be normal in Wilson disease during pregnancy or with oral contraceptive pills. The 24-hour urine copper studies are also helpful but can be falsely elevated in individuals with marked inflammatory liver disease from many different causes.
Gender affects the disease course, with women more commonly having liver disease than men.5 Girls are also more likely to present with fulminant liver failure than boys.6
Histologic Findings
As an overview, the histologic findings are variable but tend to fall into several broad categories. In terms of clinical clues, an increased suspicion of Wilson disease is useful when (1) young patients have unexplained chronic liver disease, (2) young or middle-aged individuals have both liver and neurologic disease, and (3) young individuals present with acute liver failure. Histologic clues that raise suspicion for Wilson disease include fatty liver in normoweight, younger individuals; cryptogenic cirrhosis in a young or middle-aged adult; or an acute unexplained hepatitis in a young individual.
“ALMOST NORMAL” LIVER PATTERN. In this pattern, the biopsy shows minimal nonspecific findings (eFig. 16.4) with minimal lymphocytic inflammation, minimal to absent steatosis, and often scattered glycogenated hepatocyte nuclei.
ACUTE HEPATITIS PATTERN. The clinical presentation of acute liver failure can manifest histologically as marked inflammation with hepatocyte necrosis. Plasma cells and interface activity can be prominent, and the histologic findings can mimic autoimmune hepatitis. Preexisting fibrosis can also be present.
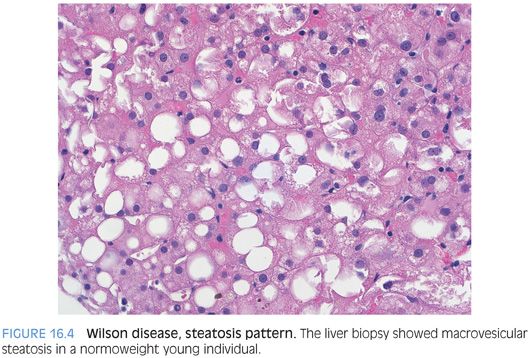
FATTY LIVER DISEASE PATTERN. In this pattern, the biopsy shows macrovesicular steatosis that can vary from mild to marked (Fig. 16.4). Mallory bodies and balloon cells can occasionally be present, and the findings can overlap substantially with fatty liver disease from the metabolic syndrome.
CRYPTOGENIC CIRRHOSIS PATTERN. In this pattern, the biopsies can show advanced fibrosis or established cirrhosis with minimal or mild septal and portal chronic inflammation. Lobular cholestasis can be seen, but most have little ongoing lobular inflammation. Steatosis can also be present. Ballooned hepatocytes and Mallory hyaline can be prominent in a subset of cases. Giant cell transformation of hepatocytes can also be occasionally seen, especially in the setting of cholestasis. If there is sufficient copper accumulation, granular reddish-brown deposits can be seen in periseptal hepatocytes (eFig. 16.5).
Copper Stains
The rhodanine copper stain is the most commonly used stain and shows a red-brown granular staining (eFigs. 16.6 and 16.7). The Timm silver sulfide stain is reported to be more sensitive7 but is not widely used. If your laboratory uses the Timm method, a longer (24 hours) deparaffination time has been recommended.8 Orcein and Victoria blue stains detect copper-binding protein and not copper itself.
Positive copper stains tend to have a zone 1 distribution,9 but the copper deposition can be panlobular. One potential pitfall when interpreting the copper stain is mistaking lipofuscin pigment for copper staining. The staining quality of a light rhodanine stain can sometimes mimic lipofuscin. If in doubt, the location of the pigment can be helpful (periportal vs. pericentral) as well as a Fontana-Masson stain (positive in lipofuscin). Positive copper stains also have to be interpreted in the context of other laboratory and histologic findings because chronic cholestasis from any cause can lead to copper accumulation, if the cholestasis is of long enough duration (typically many months to years). However, in most cases of chronic cholestasis, the copper deposition tends to be patchy and mild.
Copper stains can be very useful when evaluating for Wilson disease (see eFig. 16.6), but a negative copper stain should be interpreted cautiously if there are other clinical or laboratory findings that strongly suggest Wilson disease. In these cases, submitting the tissue for quantitative copper analysis can be very important because the copper levels can be elevated in tissue despite the negative rhodanine staining.
Iron Stains
A subset of cases with Wilson disease will also have iron accumulation in the hepatocytes, with iron levels that range from mild to marked. Iron positivity is more common in males, and iron levels can, in some cases, increase after therapy to reduce copper tissue levels.10
OTHER COPPER OVERLOAD DISEASES
There are a number of inherited copper overload diseases outside of Wilson disease. Although these diseases are rare, the most common are Indian childhood cirrhosis, Tyrolean infantile cirrhosis, and idiopathic copper toxiocosis. These diseases develop cirrhosis, often very rapidly, and cirrhosis is usually present at first diagnosis. For this reason, the precirrhotic histologic findings are poorly understood. The livers have marked copper accumulation, but genetic testing shows a lack of mutations in the ATPB7 gene. The histology for all of these three conditions is similar, and idiopathic copper toxiocosis is described in more detail as a representative histology.
Individuals with idiopathic copper toxiocosis have normal ceruloplasmin levels, in contrast to Wilson disease. Most cases present before the age of 2 years with histories of progressive lethargy, increased infections, and hepatomegaly. However, a second peak can be seen around 5 years of age and rare cases can be diagnosed as late as 10 years of age.11 The etiology remains unclear, but there may be both environmental and genetic risk factors.11,12 Essentially, all cases to date have been diagnosed at the cirrhotic stage, and the cirrhosis pattern is often that of very tiny cirrhotic nodules, termed micronodules. The hepatocytes show marked reactive changes with abundant Mallory hyaline and scattered acidophil bodies.12 Inflammation tends to be mild but can be composed of both lymphocytes and neutrophils. Cholestasis can be prominent. A subset of cases has been described as having significant fibrosis of the central veins. In addition to the micronodular pattern of cirrhosis, marked pericellular fibrosis can also be seen on trichrome.
GLYCOGEN STORAGE DISEASES
There are many glycogen storage diseases that present as abnormal accumulations of glucose within the hepatocytes.13,14 These include primarily types Ia/b, IIIa/b, VI, IX, and XI. The hepatocytes in glycogen storage diseases tend to have patterns that include either steatosis or glycogenosis or mixed pattern with both glycogenosis and steatosis (Table 16.1). PAS stains can show abundant glycogen accumulation, but this finding is not specific because even a normal liver can have abundant hepatocyte glycogen. Thus, the best approach is based on combining the H&E findings with the clinical history to suggest a diagnosis of a storage disease. In most cases, a precise diagnosis of the subtype of glycogen storage disease cannot be reliably made on the basis of histology alone. Instead, the biopsy findings are useful to demonstrate the abnormal accumulation of glycogen or fat and help refine the diagnosis. Additional biochemical assays are then needed to precisely classify the type of glycogen storage disease.14 Clinical features may also be helpful in classifying the glycogen storage disease in some cases, but the degree of clinical overlap and the frequent presence of significant clinical ambiguities limit this approach and precise biochemical assays are typically needed. Common clinical findings at presentation for many of the different forms of glycogen storage disease include hepatomegaly, hypoglycemia, short stature, and recurrent infections. The types of glycogen storage disease that are typically not associated with hypoglycemia at presentation include types II and IV. The types most likely to be associated with liver fibrosis are types III and IV, but fibrosis can also be seen in types I and IX.15










