CHAPTER 75 Wilson Disease
Copper, a component of several essential enzymes, is toxic to tissues when present in excess. Dietary intake of copper generally exceeds the trace amount required, and mechanisms to control influx and efflux from cells must maintain an appropriate balance. Two disorders of copper transport are known: Menkes disease, an X-linked defect in transport of copper from the intestine, and Wilson disease, an autosomal recessive disorder of copper overload. Wilson disease was first described, in 1912 by Kinnear Wilson, as a familial disease characterized by progressive, lethal neurologic dysfunction with chronic liver disease and a corneal abnormality, the Kayser-Fleischer (KF) ring.1 Wilson also observed that some younger siblings of typical patients died of severe liver disease without experiencing neurologic abnormalities. In this disease, inadequate hepatic copper excretion leads to copper accumulation in the liver, brain, kidney, and cornea. The incidence in most populations is on the order of 1 in 30,000.
THE COPPER PATHWAY
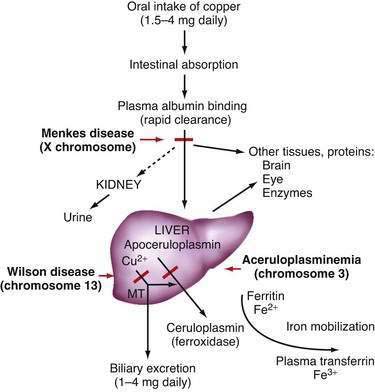
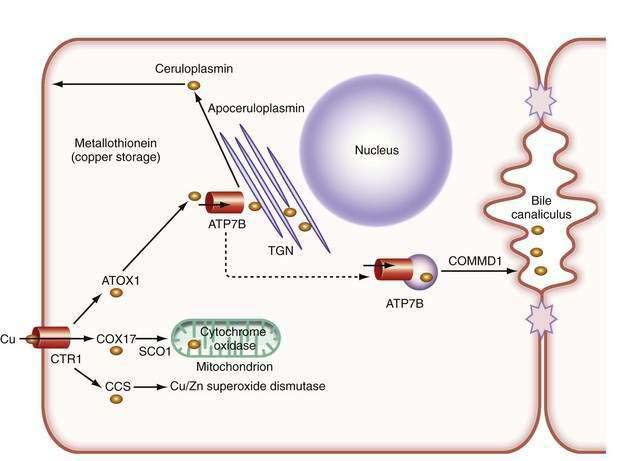
In the liver, copper is incorporated into the protein apoceruloplasmin to produce ceruloplasmin (also called holoceruloplasmin). More than 90% of the copper in plasma is an integral part of ceruloplasmin, an α2-glycoprotein that contains six molecules of copper and has a molecular weight of 132 kd. The normal serum concentration of ceruloplasmin in adults, as measured by immunochemical or enzymatic techniques, is 200 to 400 mg/L, rising from a very low level at birth to 300 to 500 mg/L in the first years of life. Ceruloplasmin is an acute-phase reactant that is elevated by inflammation (including inflammatory hepatic disease), pregnancy, and the use of exogenous estrogen. The majority of ingested copper is excreted via the bile; a small fraction is excreted in urine. When intestinal or liver cells are overloaded with copper, the metallothioneins, a class of low-molecular-weight cysteine-rich proteins, are induced and sequester copper in a nontoxic form. The normal pathways of copper transport in the body and in the hepatocyte are shown in Figures 75-1 and 75-2.
THE BASIC MOLECULAR DEFECT
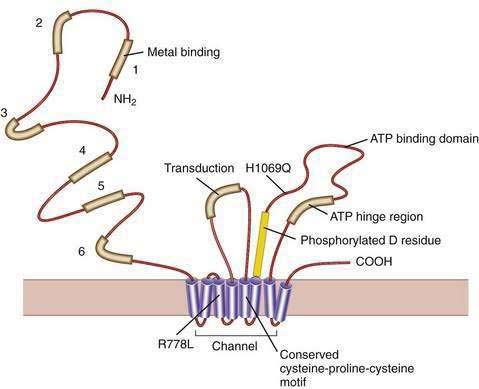
Our understanding of the basic defect in Wilson disease increased dramatically with the cloning of the genes, first for X-linked Menkes disease and then for Wilson disease. The gene for Menkes disease (ATP7A), which was cloned by using a chromosomal breakpoint in an affected female patient, was found to be related to bacterial copper resistance genes. Cloning of the Wilson disease gene (ATP7B) was accomplished by a combination of linkage analysis, physical mapping of the relevant region of chromosome 13q14, and recognition of its extensive homology with the Menkes disease gene.2,3 The coding region of the gene is 4.1 kilobases in length, with messenger RNA (mRNA) of about 8 kilobases. The product, ATP7B (or the Wilson ATPase), is a membrane P-type adenosine triphosphatase (ATPase) that consists of 1443 amino acid residues and has a molecular mass of 160 kd. The predicted structure,2 as confirmed by structural studies,4 has six copper binding domains, a phosphorylation domain, an ATP-binding region, and eight transmembrane domains. (Fig. 75-3). All functionally important regions of the gene are conserved between bacteria and yeast. Mutations in the ATP7B gene result in retention of copper in the liver as well as impaired incorporation of copper into ceruloplasmin. The Long-Evans cinnamon (LEC) rat and the toxic milk (tx) mouse have mutations in their homologous ATP7B genes and are thus suitable models for the study of Wilson disease mechanisms and therapy.5,6
Although the gene for Menkes disease is expressed in many tissues, including muscle, kidney, heart, and intestine, the gene for Wilson disease is expressed predominantly in the liver and kidney, with minor expression in brain, lungs, and placenta.2 Studies in cultured cells show localization of the Wilson ATPase in the trans-Golgi network, with trafficking from the trans-Golgi network to cytoplasmic vesicles in the presence of increased copper.7 When intracellular copper concentrations are elevated, the Wilson ATPase is found near the apical (bile canalicular) membrane in hepatocytes, consistent with its proposed function of facilitating excretion of copper via bile.8
Additional proteins are involved in the intracellular transport of copper. Molecular copper is not free in the cell but is transported to specific proteins by copper chaperones.9 The chaperone ATOX1 transports copper to the Wilson ATPase. Other chaperones deliver copper to superoxide dismutase in the cytoplasm and various copper-containing proteins in mitochondria.
The study of inherited hepatic copper toxicosis in Bedlington terriers has identified a new component of the copper transport system. Affected dogs show clinical variability that ranges from death or hepatic disease at two to three years of age to less severe chronic disease to a high level of hepatic copper. The proposed defective canine gene was identified by positional cloning through the use of markers to identify a region containing the COMMD1 (initially called the MURR1) gene. The gene has a deletion of one exon in some,10 but not all,11 affected dogs. Although the gene responsible for the excess copper remains to be identified, the disease in Bedlington terriers has highlighted other genes that may be involved in response to excess copper; COMMD1 interacts not only with the Wilson ATPase,12 but also with the X-linked inhibitor of apoptosis (XIAP), which can also bind copper.13 COMMD1 could be involved in response to, and facilitate elimination of, excess copper.
CLINICAL FEATURES
The clinical presentation of Wilson disease is extremely variable. The age at onset of symptoms is much broader than previously thought, ranging from 3 to 55 years. Wilson disease with hepatic involvement has been identified in one- to two-year-old patients and in patients older than age 60.14 Patients may present with chronic or fulminant liver disease, a progressive neurologic disorder without clinically prominent hepatic dysfunction, isolated acute hemolysis, or psychiatric illness. The clinical variability often makes confirmation of the diagnosis difficult. Further description of the clinical features and treatment has been published.14,15
HEPATIC PRESENTATION
Wilson disease may present in children and young adults with clinical liver disease indistinguishable from autoimmune hepatitis.16 As in autoimmune hepatitis, the onset may be acute. Fatigue, malaise, arthropathy, and rashes may occur; laboratory findings include elevated aminotransferase levels, a greatly increased serum immunoglobulin (IgG) concentration, and detectable nonspecific autoantibodies such as antinuclear antibodies and anti-smooth muscle (anti-actin) antibodies. Wilson disease must be specifically ruled out because the treatment of the two diseases is entirely different. With appropriate treatment, the long-term outlook for patients with Wilson disease that manifests as autoimmune hepatitis appears to be favorable, even if cirrhosis is present.
Wilson disease may present as fulminant hepatic failure, with severe coagulopathy and encephalopathy. Acute intravascular hemolysis is usually present, and renal failure may develop. Because the patient typically has not been suspected of having underlying liver disease, fulminant viral hepatitis is usually the working diagnosis. Unlike fulminant viral hepatitis, fulminant Wilson disease is typically characterized by disproportionately low aminotransferase levels (usually much less than 1500 U/L) at the onset of clinically apparent disease. The serum alkaline phosphatase level is in the normal range or even low for the patient’s age, and the bilirubin level is often disproportionately elevated as a result of hemolysis. In adults who present with Wilsonian fulminant liver failure, the combination of a ratio of the alkaline phosphatase level to the total bilirubin level of less than 4 and a ratio of the aspartate aminotransferase (AST) to alanine aminotransferase (ALT) level of greater than 2.2 can be extremely helpful for making the diagnosis, whereas the serum ceruloplasmin level is not (see later).17 Slit-lamp examination may reveal Kayser-Fleischer rings (see later). Urinary copper excretion is greatly elevated. These patients require urgent liver transplantation because they do not respond well to chelation treatment; albumin dialysis and related techniques may serve as temporary procedures until liver transplantation can be performed (see later).18 This presentation of Wilson disease is not rare, and affected patients account for a substantial proportion of persons transplanted for fulminant hepatic failure (see Chapters 93 and 95).
Recurrent bouts of hemolysis may predispose to the development of gallstones. Cirrhosis, if present, may be a further predisposing factor. Children with unexplained cholelithiasis, particularly with small bilirubinate stones, should be tested for Wilson disease. Compared with other chronic liver diseases, Wilson disease is rarely complicated by hepatocellular carcinoma; however, patients may have an increased propensity to abdominal malignancies,19 and hepatocellular carcinoma has been reported in a child with Wilson disease.
PATHOLOGY
Changes in hepatocellular mitochondria, identified with electron microscopy, are an important feature in Wilson disease.20 The mitochondria vary in size, and the numbers of dense bodies in mitochondria may be increased. The most striking change is dilatation of the tips of the mitochondrial cristae as a result of separation of the inner and outer membranes of the cristae, with widening of the intercristal space until the appearance is irregularly cystic. The crista resembles a tennis racquet if only the tip is dilated. This finding, although not entirely specific for Wilson disease, can be helpful diagnostically, even in young and minimally affected patients. Involvement of hepatocytes may be not uniform, and abnormalities may be found in some hepatocytes in some lobules and not in others. The mitochondrial changes are probably a consequence of oxidative damage from excessive copper in hepatocytes.21
DIAGNOSIS
The patient with the classic combination of chronic liver disease, tremor or dystonia, and Kayser-Fleischer rings is readily diagnosed on clinical grounds, but such patients are uncommon. A diagnostic scoring system has been proposed22 but has been evaluated only in children. Suggestive clinical symptoms are often the main prerequisite for diagnosing Wilson disease, and laboratory investigations may provide confirmation. Kayser-Fleischer rings should be sought through a careful slit-lamp examination and repeated if necessary. Lack of Kayser-Fleischer rings does not exclude the diagnosis of Wilson disease. Routine liver biochemical test levels are usually abnormal, with mild-to-moderate elevations of the serum aminotransferase levels. Serum levels of the ALT may be much lower than those of AST, possibly reflecting damage to hepatocellular mitochondria.
Two major disturbances of copper disposition in Wilson disease are defective incorporation of copper into ceruloplasmin and decreased biliary excretion of copper. A summary of biochemical features in Wilson disease in comparison with normal persons is shown in Table 75-1






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


