CHAPTER 83 Vascular Diseases of the Liver
Vascular disorders of the liver are relatively uncommon but frequently result in serious liver disease and portal hypertension.1,2 The continuously high metabolic activity of the liver makes it particularly susceptible to vascular compromise; however, its complex dual blood supply offers unique protection against ischemic injury. Hypercoagulable states play an important role in the pathogenesis of many of these disorders, and knowledge of them is essential for understanding and treating the associated hepatic vascular conditions. This chapter reviews a heterogeneous group of disorders resulting from hepatic vascular and cardiovascular diseases. Vasculitis involving the liver is discussed in Chapter 35.
BUDD-CHIARI SYNDROME
Hepatic venous outflow obstruction is the hallmark of the Budd-Chiari syndrome. Reductions in hepatic venous outflow can occur anywhere from the right atrium to the small hepatic venules and result in dramatic anatomic and physiologic changes. Classic Budd-Chiari syndrome results from thrombosis of one or more hepatic veins at their openings into the inferior vena cava. The deleterious physiologic changes of hepatic venous obstruction are transmitted directly to the hepatic sinusoids, resulting in sinusoidal congestion, portal vein hypertension, and reduced portal vein blood flow. The result is hepatomegaly, pain, ascites, and impaired hepatic function. The ascitic fluid typically has a high serum-ascites albumin gradient and a high protein content as a result of the increased filtration of serum proteins through the highly permeable sinusoidal spaces (see Chapter 91). The progression of disease is rarely fulminant; in most patients, the clinical course is subacute and less than six months in duration.1 In older series, the mortality rate in untreated cases was as high as 90% at three and one half years.3 With advances in diagnostic imaging and improved medical, surgical, and radiologic treatments for Budd-Chiari syndrome, however, survival has improved significantly since 1980.
The literature on Budd-Chiari syndrome is extensive. Two major reviews of the world literature collected data on cases reported before 1980.4,5 Reviews of more than 100 cases have appeared from Japan, India, China, and South Africa.6–9 The review from India is especially helpful in demonstrating the geographic diversity of this syndrome.7
ETIOLOGY
Anatomically, Budd-Chiari syndrome results from hepatic vein obstruction, inferior vena cava obstruction (above or at the level of the hepatic veins), or both. The main causes of the syndrome are listed in Table 83-1. In Western countries, thrombosis of the hepatic veins is the most common presentation, whereas in Asia and Africa, membranous obstruction of the inferior vena cava (MOVC) accounts for more than 40% of cases.10 Thrombogenic states can be identified in at least 75% of cases not caused by MOVC. Increasingly sophisticated testing for hypercoagulable states has reduced the frequency of idiopathic cases to less than 10%.1,2,11 Hematologic disorders are the most common causes of Budd-Chiari syndrome. Primary myeloproliferative diseases, particularly polycythemia vera, may account for 50% of cases.1,2 In addition, latent myeloproliferative disorders may be detected using cell culture techniques12 or by testing for mutations (specifically the V617F mutation) in the gene coding for the tyrosine kinase Janus kinase 2 (JAK2).13 Testing for JAK2 mutations is likely to play an increasing role in the evaluation of patients with Budd-Chiari syndrome and a suspected myeloproliferative disorder. Tumors, infections, and pregnancy each account for about 10% of cases. Other hypercoagulable states associated with Budd-Chiari syndrome include paroxysmal nocturnal hemoglobinuria, antiphospholipid syndrome,14 and deficiencies of antithrombin, protein C, and protein S.15 More recently recognized causes include factor V Leiden mutation16 and mutations of the prothrombin gene and the methylenetetrahydrofolate reductase gene.17 Oral contraceptive use increases the risk of Budd-Chiari syndrome by more than two-fold, especially in the presence of other hypercoagulable states. More than 25% of patients are found to have multiple thrombogenic risk factors.17
Table 83-1 Causes of Budd-Chiari Syndrome
| Hypercoagulable States |
| Infections |
| Malignancies |
| Miscellaneous |
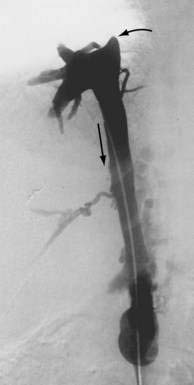
The pathophysiologic characteristics of MOVC are poorly understood. The disorder is much more common in developing countries, especially Asia and Africa, than in Western countries. More than 70% of cases of Budd-Chiari syndrome in China are due to MOVC. In India the incidence of MOVC, in contrast to classic Budd-Chiari syndrome, seems to be decreasing.18 The clinical presentation usually is subacute or chronic. The membranous webs may be thick or thin and typically occur in the intrahepatic inferior vena cava, often with occlusion of the ostia of the hepatic veins (Fig. 83-1). A congenital origin for the lesion has been proposed on the basis of the complex embryologic development of the inferior vena cava and reported presentations in childhood. An acquired origin, however, is supported by the peak occurrence in the fourth decade of life and histologic studies suggesting that the membrane develops from an organizing thrombus. Hypercoagulability is relatively less common in MOVC than in hepatic vein thrombosis. Therefore, other explanations for thrombosis in MOVC have been proposed, such as chronic infection, endothelial trauma caused by movement of the diaphragm with respiration and coughing, and venous turbulence resulting from the right-angle flow of blood from the hepatic veins into the inferior vena cava. Another feature of MOVC is the propensity of affected persons to develop hepatocellular carcinoma, which is less common in classic Budd-Chiari syndrome.19,20 The distinctive features of MOVC have led some investigators to consider MOVC a separate clinical entity termed obliterative hepatocavopathy.21
CLINICAL FEATURES
The epidemiologic characteristics of Budd-Chiari syndrome (except those cases associated with MOVC) parallel those of its underlying conditions. The syndrome is rare in infants and young children; the largest pediatric series describes South African children with MOVC. More than one half of the cases of classic Budd-Chiari syndrome occur between the ages of 20 and 39 years.5 Budd-Chiari syndrome occasionally may be identified in asymptomatic persons undergoing evaluation for mildly elevated liver biochemical test levels.22 In these patients, the lack of symptoms probably is the result of thrombosis of only one hepatic vein or the development of large venous collaterals.
PATHOLOGY
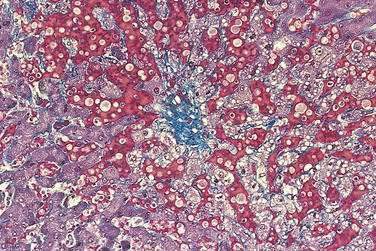
Acutely, the hepatic histologic features of centrilobular congestion, hemorrhage, sinusoidal dilatation, and noninflammatory cell necrosis predominate (Fig. 83-2). Within weeks, fibrosis develops in the centrilobular areas, more so than in periportal areas. Over time, these lesions evolve into cirrhosis. Large regenerative nodules are common, especially in areas of decreased portal venous perfusion.23 Indeed, portal vein thrombosis can be seen in 10% of cases and in 50% of hepatic explants at the time of liver transplantation for Budd-Chiari syndrome.24 Because hepatic vein occlusion is asymmetrical, the pathologic effects may vary in different regions of the liver. Massive caudate lobe hypertrophy is a common feature, probably because of preservation of venous drainage directly into the inferior vena cava, and may contribute to compression of the inferior vena cava. Pathologic changes (except for cirrhosis) can be reversed with adequate decompression of the hepatic sinusoids (see later).
DIAGNOSIS
Doppler ultrasonography, with sensitivity and specificity rates greater than 80%, is the diagnostic procedure of first choice.25 It is relatively inexpensive, safe, and available in most hospitals. Typical Doppler ultrasonographic features of Budd-Chiari syndrome include lack of visualization of normal hepatic venous connections to the inferior vena cava, comma-shaped intrahepatic or subcapsular collateral vessels, and absence of flow signal in the hepatic veins. The diagnostic accuracy of ultrasonography is decreased by a large body habitus and is operator dependent.
Magnetic resonance imaging (MRI) and computed tomography (CT)26 may also demonstrate characteristic features of Budd-Chiari syndrome but do not add much to the findings of an adequate ultrasonographic examination.27 MRI may be a better second-line test than CT because of the ability to provide accurate angiographic detail of the hepatic vein and inferior vena cava anatomy with minimal risk of nephrotoxicity. The combination of Doppler ultrasonography and either MRI or CT imaging should be sufficient to diagnose most cases of Budd-Chiari syndrome.
For years, venography was the standard for diagnosis of Budd-Chiari syndrome; however, with improvements in less invasive radiologic imaging, venography is often unnecessary for diagnostic purposes alone. Venography should be performed in cases of suspected Budd-Chiari syndrome when first- and second-line imaging tests are nondiagnostic and when surgery and other therapeutic interventions are planned. Measurement of the hepatic venous pressure gradient is required when vena cava stenosis is present, to plan for portosystemic shunt surgery (see Chapter 90). Venography also allows access for transjugular biopsy of both the right and left hepatic lobes, to confirm the diagnosis of Budd-Chiari syndrome and guide therapy. Liver biopsy is not essential for making the diagnosis of Budd-Chiari syndrome, however, and clinical staging of hepatic synthetic function (e.g., Child-Turcotte-Pugh score) may be better for planning treatment (see later).28
TREATMENT
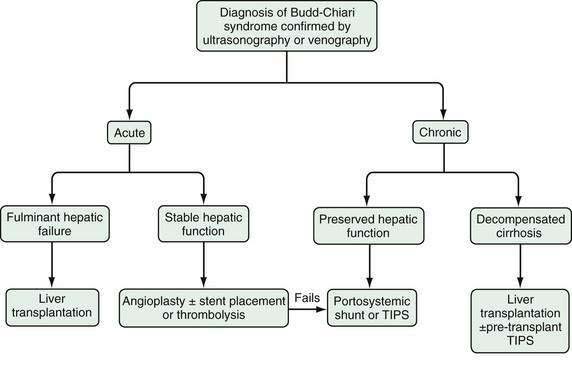
The therapy of Budd-Chiari syndrome depends on the etiology, anatomic characteristics, and pace of the disease (Fig. 83-3). The precipitating causes of Budd-Chiari syndrome must be evaluated and treated. Most patients need some form of intervention, and collaboration among a hepatologist, interventional radiologist, and hepatobiliary surgeon is optimal. Treatment options consist primarily of combinations of medical therapy with diuretics and anticoagulants, interventional therapy to decompress the hepatic sinusoids and prevent further hepatic necrosis, and liver transplantation for hepatic failure.
Medical management alone may be appropriate for the few patients with milder forms of Budd-Chiari syndrome. Treatment typically consists of diuretic therapy with spironolactone and furosemide and dietary sodium restriction to achieve a negative sodium balance (see Chapter 91). Large-volume paracentesis may be needed to relieve tense ascites. Anticoagulation is recommended and consists of intravenous heparin followed by warfarin to achieve an international normalized ratio (INR) for prothrombin of 2.0 to 2.5. Medical therapy is considered successful if ascites is controlled, liver biochemical test results improve or normalize, and symptoms resolve. Most patients with Budd-Chiari syndrome do not respond adequately to medical therapy alone, however, and require some form of intervention to decompress the hepatic sinusoids.
Many case reports and small series have described the use of thrombolytic therapy for acute Budd-Chiari syndrome. Thrombolytic agents are most effective if administered within three weeks of the onset of symptoms, if flow is demonstrated in the thrombosed vein, and if the agent is infused directly into the occluded hepatic vein. Systemic and hepatic arterial infusions are less effective.29 Angioplasty is often performed in conjunction with thrombolysis to improve vein patency and to reopen an acutely thrombosed stent or transjugular intrahepatic portosystemic shunt (TIPS) (see later).
The role of interventional radiology in the treatment of Budd-Chiari syndrome has expanded greatly. Angioplasty, with or without placement of a stent through short and localized stenoses of the hepatic veins or the inferior vena cava, can relieve symptoms in more than 80% of patients with MOVC and is the primary treatment for this disorder in many parts of the world.30–33 The rate of restenosis is high, and regular follow-up with Doppler ultrasonography is required.34 Angioplasty can also be combined successfully with surgical creation of a portacaval shunt in patients with both inferior vena cava and hepatic vein obstruction (see later).35
Use of TIPS has gained popularity as a treatment for refractory complications of portal hypertension (see Chapter 90). Procedural mortality is low, and shunt placement is usually successful,36 even in complete hepatic vein occlusion, in which a shunt can be placed between the intrahepatic vena cava and portal vein.37 TIPS is also useful for treating combined hepatic vein and inferior vena cava obstruction and can be effective in patients with fulminant Budd-Chiari syndrome awaiting liver transplantation. Short-term mortality rates in the latter setting may be as high as 50%38; however, long-term clinical stabilization after TIPS placement without liver transplantation also has been reported.39 TIPS has been used successfully in patients with acute Budd-Chiari syndrome in whom thrombolytic therapy and angioplasty have failed. Finally, TIPS is an effective “bridge” to liver transplantation in patients with chronic Budd-Chiari syndrome who have liver failure and refractory ascites or variceal bleeding. Unfortunately, TIPS dysfunction requiring revision occurs in as many as 70% of patients40 and seems to be more common in patients with Budd-Chiari syndrome than in those with other indications for TIPS placement. Polytetrafluoroethylene-covered stents significantly reduce rates of TIPS dysfunction and the need for reintervention and improve primary patency rates at one year from 19% to 67%.41 TIPS placement that extends too far into the portal vein or into the suprahepatic vena cava makes liver transplantation more complicated.
In Western countries, surgical therapy for Budd-Chiari syndrome consists mainly of portosystemic shunting and liver transplantation. Surgical therapy of MOVC that is unsuitable for or refractory to angioplasty and stent placement may entail resection or transatrial “finger fracture” of vena cava webs42 or dorsocranial liver resection with a hepaticoatrial anastomosis (the Senning procedure).43 For classic Budd-Chiari syndrome, portosystemic shunting relieves portal hypertension effectively, thereby alleviating hepatic ischemic necrosis, refractory ascites, and variceal bleeding. When shunt surgery is successful, the portal vein becomes the hepatic outflow tract, hepatomegaly resolves, hepatic histologic findings improve and may even normalize, and survival is prolonged in more than 90% of cases.44
The choice of a portosystemic shunt depends on the degree of hepatomegaly and caudate lobe hypertrophy, presence or absence of inferior vena cava stenosis, and surgical expertise. Portacaval and mesocaval shunts are associated with the best results. With mesocaval shunts, the rate of shunt thrombosis is higher—33% at five years45—but placement is technically simpler when portal dissection is impeded because of caudate lobe hypertrophy. Thrombosis of portacaval shunts is uncommon, with a rate of only 3% over 13 years,44 but portacaval shunts increase the technical difficulty of subsequent liver transplantation more than do mesocaval shunts. Dysfunction of a surgical portosystemic shunt significantly reduces long-term survival and is more likely with prosthetic grafts or with high portal venous pressures.46 Placement of a portacaval or mesocaval shunt is contraindicated if inferior vena cava stenosis is present and the vena cava pressure is greater than 20 mm Hg or if the portacaval pressure gradient is less than 10 mm Hg. Surgical options in these patients include placement of a mesoatrial shunt,47,48 a combined portacaval and cavoatrial shunt,44 and combinations of surgical shunt creation with vena cava angioplasty and stent placement.35 Because of high rates of shunt thrombosis, however, TIPS may be the best option in these patients.
Liver transplantation is appropriate in patients with liver failure resulting from fulminant or chronic Budd-Chiari syndrome and in patients with a failed surgical portosystemic shunt (see Chapter 95).49 In patients with protein C, protein S, or antithrombin deficiency, liver transplantation also cures the underlying hypercoagulable state, although most patients will require lifelong anticoagulation. Underlying myeloproliferative disorders can be managed with hydroxyurea and aspirin after liver transplantation,50 but there is a risk of progression and leukemic transformation.51 Recurrent Budd-Chiari syndrome after liver transplantation occurs in 4% to 10% of patients, and the risk of thrombosis of the hepatic artery and portal vein is increased as well.52,53 In addition, bleeding complications are more common because of anticoagulant therapy.54 Despite these drawbacks, five-year survival rates for patients with Budd-Chiari syndrome who undergo liver transplantation exceed 85%.53
The choices of therapy in patients with Budd-Chiari syndrome are complicated, in great part because of the lack of large, controlled studies comparing various treatments and the lack of standardization in classifying Budd-Chiari syndrome into acute, subacute, and chronic forms.55 One multivariate analysis failed to show a survival advantage in surgically shunted patients compared with those receiving medical treatment alone.56 Although medium-term studies of TIPS as definitive therapy in Budd-Chiari syndrome have been reported, there are no long-term controlled studies comparing TIPS with surgical therapy. TIPS is clearly an effective and widely available therapy; however, shunt dysfunction is common and leads to frequent reinterventions. On the other hand, surgical shunts are associated with significant perioperative morbidity and mortality, but when performed successfully, they offer good long-term outcomes. Surgical expertise in complex shunt surgery, however, is declining with the increasing use of TIPS. Surgical shunt surgery seems to be most efficacious in patients with acute or chronic Budd-Chiari syndrome with refractory symptoms and preserved hepatic synthetic function (Child classes A and B [see Chapter 90]).57 Patients with fulminant or chronic Budd-Chiari syndrome with impaired hepatic synthetic function (Child classes B and C) should be considered for liver transplantation, with or without prior TIPS placement. (As compared with the Child class, the Model for End-stage Liver Disease (MELD) score does not incorporate an assessment of refractory ascites and is less useful for estimating prognosis in patients with Budd-Chiari syndrome than in those with other causes of advanced chronic liver disease [see Chapter 95]).
In patients with acute Budd-Chiari syndrome, attempts should be made to decompress the hepatic sinusoids with combinations of angioplasty, stent placement, and thrombolytic therapy, followed by TIPS placement or creation of a surgical portosystemic shunt in those in whom these treatment approaches fail. A proposed stepwise (“minimally invasive”) approach beginning with anticoagulation therapy, then attempted hepatic vein recanalization, followed by TIPS, and then liver transplantation in treatment failures has been reported to achieve an overall five-year survival rate of nearly 90%.58
SINUSOIDAL OBSTRUCTION SYNDROME (VENO-OCCLUSIVE DISEASE)
Occlusion of the terminal hepatic venules and hepatic sinusoids resembles the Budd-Chiari syndrome clinically; however, the causes, epidemiologic and pathophysiologic characteristics, and prognosis of this entity are sufficiently distinct to justify a separate designation. In the past, the entity was known as veno-occlusive disease. Because the occlusion consistently involves the hepatic sinusoids, the term sinusoidal obstruction syndrome has been proposed as a more appropriate name for this disorder.59
ETIOLOGY
Liver disease caused by Senecio poisoning was originally reported from South Africa in 1920 (see Chapter 87),60 but the term veno-occlusive disease was not used until 1954, when it was related to the ingestion of pyrrolizidine alkaloids contained in plants of the genera Senecio, Crotalaria, and Heliotropium in Jamaica.61 Ingestion of alkaloids in inadequately winnowed wheat or in “bush tea,” especially in malnourished persons, is the main cause of veno-occlusive disease (sinusoidal obstruction syndrome) worldwide. Epidemics have been reported in India, Afghanistan, South Africa, the Middle East, and the United States. More recently, the herbal remedy comfrey (genus Symphytum) has been associated with sinusoidal obstruction syndrome. Rare familial clusters have been reported in association with immunodeficiency states.62
Since the advent of cancer chemotherapy in the 1950s, sinusoidal obstruction syndrome in Western countries has occurred most commonly after bone marrow transplantation (see Chapter 34).63 A variety of antineoplastic drugs have been implicated as causes of sinusoidal obstruction syndrome including gemtuzumab ozogamicin, actinomycin D, dacarbazine, cytosine arabinoside, mithramycin, and 6-thioguanine. Hepatic irradiation and therapy with busulfan plus cyclophosphamide also are established risk factors (see Chapters 34 and 39). In addition, long-term immunosuppression with azathioprine and 6-thioguanine used in patients with inflammatory bowel disease and in kidney and liver transplant recipients has been reported to cause sinusoidal obstruction syndrome.63
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


