Transport defects and enzymatic deficiencies
Disaccharidase deficiency
Sodium–hydrogen exchanger (congenital sodium diarrhea)
Chloride–bicarbonate exchanger (chloride- losing diarrhea)
Sodium–glucose cotransporter (glucose–galactose malabsorption)
Lysinuric protein intolerance
Chylomicron retention disease
Abetalipoproteinemia
Ileal bile acid receptor defect
Enterokinase deficiency
Inherited epithelial defects and villous atrophy
Autoimmune enteropathy
IPEX syndrome
Microvillus inclusion disease
Tufting enteropathy
Endocrine cell dysgenesis
Other
Coeliac disease
Allergic enteropathy
Eosinophilic enteritis
Infectious/ post-infectious enteropathy
Lymphangiectasia
Acrodermatitis enteropathica
Metabolic diseases
Tumours
Idiopathic
The most frequent diagnosis in children with protracted diarrhea is autoimmune enteropathy (AIE) [3, 4]. It is a rare, immune-mediated disorder starting usually within the first months of life. The age of onset is between 1 month and 5 years (median age 17 months) [5], but late-onset adult forms have been also reported [6–9]. The disease was first described by Walker-Smith et al. in 1982 in a male child with clinical features of coeliac disease and villous blunting unresponsive to gluten-free diet [10] and represents a heterogeneous group of disorders rather than a discrete entity. The incidence is estimated at less than 1 in 100,000 infants. The diagnostic criteria are debatable but the presence of circulating anti-enterocyte antibodies and the lack of immunodeficiency have been proposed as the hallmark features of AIE [5, 11]. The latter criterion has been challenged by clinical experience and better understanding of the immunology of autoimmunity and self-tolerance [12].
AIE is characterized by variable clinical expression, ranging from isolated gastrointestinal involvement to severe systemic disease [13, 14]. Patients diagnosed with the disease often exhibit extra-intestinal manifestations of autoimmunity, in contrast to those with tufting enteropathy and microvillus inclusion disease [15]. Based on a genetic approach combined with immunological evaluation, three different forms of AIE have been proposed:
1.
A predominately or isolated gastrointestinal form of AIE with typical anti-enterocyte antibodies in both sexes
2.
A systemic X-linked form of AIE associated with different endocrinopathies, haematological symptoms and severe eczematous skin disease, known as immune dysregulation, polyendocrinopathy, AIE X-linked syndrome (IPEX) occurring only in males
3.
An IPEX-like form, a priori FOXP3 independent occurring in both sexes
IPEX and autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) syndrome (APR-1/autoimmune phenomena, polyendocrinopathy, candidiasis and ectodermal dystrophy) are systemic forms of AIE [16].
Clinical Presentation
Chronic, secretory diarrhea refractory to bowel rest that leads to dehydration, malabsorption and severe weight loss is the typical clinical presentation of AIE. Diarrhea usually begins between 2 and 4 weeks of age and the secretory component can be delayed for a few months [3, 11, 17]. The symptoms are often debilitating and the disease is potentially life threatening. The establishment of the diagnosis is crucial in order to ensure optimal treatment. Patients typically require immunosuppressive therapies and total parenteral nutrition (TPN) for hydroelectrolytic balance and nutritional support [18, 19].
Despite the fact that the mucosal abnormality is primarily confined to the small intestine, the term “generalized autoimmune gut disorder” has been used to describe the association between AIE and autoimmune colitis [8]. Emerging evidence suggests that AIE can be a manifestation of a more diffuse autoimmune disorder of the gastrointestinal system, which comprises gastritis , colitis, hepatitis and pancreatitis with positivity of a variety of autoantibodies , including anti-parietal, anti-goblet cell and anti-smooth muscle antibodies [6, 20–22].
Furthermore, the involvement of extra-intestinal organs can be present during the course of the disease. Multisystem extra-intestinal manifestations include endocrine, renal, pulmonary, hematologic and musculoskeletal. Hypothyroidism with interstitial fibrosis and lymphocytic infiltration of the thyroid gland, nephrotic, nephritic syndrome and membranous glomerulonephritis, interstitial pneumonopathy, periportal fibrosis and bronchitis, haemolytic anaemia, rheumatoid arthritis and dermatitis/atopic eczema have all been reported [5, 11, 22, 23].
Pathogenesis
The underlying immunologic and molecular mechanisms in AIE have not yet been fully elucidated and are widely debatable. However, it has been established that an autoimmune response is involved in the pathogenesis of the disease. Thymus orchestrates a healthy immune system . The intrathymic maturation of T lymphocytes is crucial for the deletion of potentially self-reactive clones of T cells. The dysfunction of the thymus results in the non-deletion and presence of self-reactive T cells that can induce the expansion of anti-self B cells [11, 26, 27].
In AIE , the gut is the site where the autoimmune reaction takes place and is mediated by the activation of self-reactive T cells locally, resulting in the typical histological lesions. In normal states, the expression of human leukocyte antigen (HLA) class II molecules on the enterocyte surface is crucial in establishing and maintaining the oral tolerance as the epithelial cells present exogenous peptides to the clonotypic T cell receptors. The overexpression of HLA-DR antigens in enterocytes and the inappropriate expression of HLA class II molecules in the crypt epithelium of the proximal small intestine in children with AIE have been reported [15, 28].
An increase in the levels of cluster of differentiation CD4 and CD8 lymphocytes in the lamina propria in subjects affected by AIE provides further evidence that the T cells are involved in the pathogenesis of the disease [29, 30]. The intestinal T lymphocytes cause damage to the enterocytes by exerting direct cytotoxicity, via the production of lymphokines or through an antibody-dependent cytotoxicity resulting in cellular apoptosis [31–33]. The loss of the regulatory function of T lymphocytes and the activation of the immune system are implicated in the pathogenesis of IPEX syndrome , whereas AIE is partly attributed to a humoral immune response with the presence of anti-enterocyte antibodies [34].
A variety of circulating autoantibodies, such as antibodies against gastric parietal cells, pancreatic islets, glutamic acid decarboxylase, insulin, smooth muscle, endoplasmic reticulum, reticulin, gliadin, adrenal cells, nuclear antigens, deoxyribose nucleic acid (DNA), thyroglobulin and thyroid microsomes, has been detected in patients with AIE [7, 18]. The presence of antibodies against goblet cells, enterocytes and colonocytes is supportive of the diagnosis. These antibodies are directed against components of the intestinal brush border membrane, with an increasing intensity from the crypts towards villous tip [5, 13]. However, they are neither diagnostic nor specific for the disease and have been also identified in other disorders such as the cow’s milk allergy, inflammatory bowel disease and in adults with human immunodeficiency virus (HIV) infection . Moreover, the appearance of the autoantibodies after the onset of the mucosal damage, the lack of correlation between the titer and the histological severity and their disappearance after treatment, but before the complete mucosal restoration support the hypothesis that these antibodies are most likely a secondary event in the pathogenesis of the disease in response to bowel injury [10, 35–37].
The nature of the gut antigen that elicits the immune response and results in the alteration of the intestinal permeability has been extensively investigated. A 55-kD protein located in both the gut and renal epithelial cells that reacted with serum autoantibodies was first identified by Colletti et al. in 1991 in a patient with complicated presentation of AIE with small bowel and glomerular involvement [38]. A few years later, a 75-kD auto-antigen that is distributed through the whole intestine and the kidney was recognized in patients with X-linked AIE associated with nephropathy [39]. The intestinal auto-antigen in autoimmune polyendoendocrine syndrome type 1 (APECED) is tryptophan hydroxylase (TPH), which is mainly present in the enterochromaffin cells of the mucosa [40].
Emerging evidence has pointed towards an uncontrolled inflammatory reaction caused by the disturbance of the effector–regulatory T-cell interaction and leading to the production of autoantibodies, such as anti-enterocyte antibodies [41]. The understanding of the underlying molecular mechanism and the identification of the genetic defect in AIE was achieved due to the clinical similarities between scurfy mice and boys with the disease. Scurfy mice are a naturally occurring X-linked mutant that presents with massive lymphoproliferation, diarrhea, intestinal bleeding, scaly skin, anaemia, thrombocytopenia and hypogonadism [42]. Based on the observation that the disease-causing mutation in scurfy mice was on the X chromosome, the human IPEX locus was identified on chromosome Xp11.23-q13.3 and the gene was named FOXP3 . It comprises 11 exons which encode the FOXP3 protein or scurfin, a 48-kDa protein of the forkhead (FKH)/winged-helix transcription factor family that is predominantly expressed in CD4+CD25+ T cell with regulatory function, at significantly lower levels in CD4+CD25-T cells and not at all in CD8+ or B220+ cells [43–46].
Increasing experimental evidence has shown that scurfin is implicated in the thymic maturation of T cells that are designated to acquire regulatory function. CD4+CD25+ Treg represent a small subset (5–10 %) of CD4+ T helper cells in humans and mice. Studies on CD4+CD25+ T cells from IPEX patients with the use of anti-CD127 have shown that FOXP3 plays a crucial role in the generation of functional T regulatory cells (Treg) and intact FOXP3 is indispensable for the development of fully functional Tregs, whereas FOXP3 with amino acid substitutions in the FKH domain is sufficient for the generation of functionally immature Tregs [47].
FOXP3 has DNA-binding activity and due to its structure may serve as nuclear transcription factor and act as a repressor of transcription and regulator of T cell activation [48, 49]. The transcription of a reporter containing a multimeric-FKH-binding site is repressed by intact FOXP3. Such FKH-binding sites are located adjacent to nuclear factor of activated T cells (NFAT), regulatory sites in various cytokine promoters such as interleukin (IL)-2, or granulocyte–macrophage colony-stimulating factor enhancer. Therefore, intact scurfin protein appears to be capable to directly repress NFAT-mediated transcription of the IL-2 gene in CD4+ cells upon activation [50].
Despite the evidence that FOXP3 plays a key role in the development and function of Treg cells, the underlying mechanisms have not been fully understood. Data from animal models with transgenic induction of FOXP3 have shown that the overexpression of scurfin in normal mice leads to a tremendous suppression of immune functions, whereas the depletion of Tregs in healthy mice results rapidly in the development of different T-cell-mediated autoimmune disorders, similar to scurfy in mice or IPEX in humans that go in complete remission upon reconstitution with Treg cells [51, 52].
The three domains that are crucial for the function of FOXP3 are the C-terminal region, which contains the forkhead domain that directly binds DNA regions, the central domain with a zinc finger and leukine zipper that promotes the oligomerization of the FOXP3 molecule and the repressor domain located in the N-terminal region that binds the NAFT [53, 54]. Genetic screening on X chromosome in patients with AIE revealed that the majority of mutations cluster primarily within the FKH domain and the leukine zipper within the coding region of the FOXP3 gene causing potentially absent FOXP3 protein expression or a protein product with loss of function [13, 53].
Histopathology
Histologic evaluation of the small bowel in typical AIE reveals partial or total villous blunting/atrophy and crypt hyperplasia. In addition, there is a marked infiltration of the lamina propria by mixed inflammatory cells with a prominence of mononuclear cells, including T lymphocytes [15]. Apoptotic bodies and intraepithelial lymphocytes are present in the crypt epithelia. Most cases show a relative paucity of surface lymphocytosis in contrast to coeliac disease. The lymphocytic infiltration of the intestinal mucosa is constituted by CD4–CD8 T lymphocytes and macrophages. Goblet, Paneth and/or enterochromaffin cells may be reduced in number or absent. Cryptitis and crypt abscesses have been reported in severe AIE. Crypt enterocytes commonly show an increased expression of HLA-A, -B, -C molecules [8, 55, 56]. (See Table 2.2; Fig. 2.1) [12].
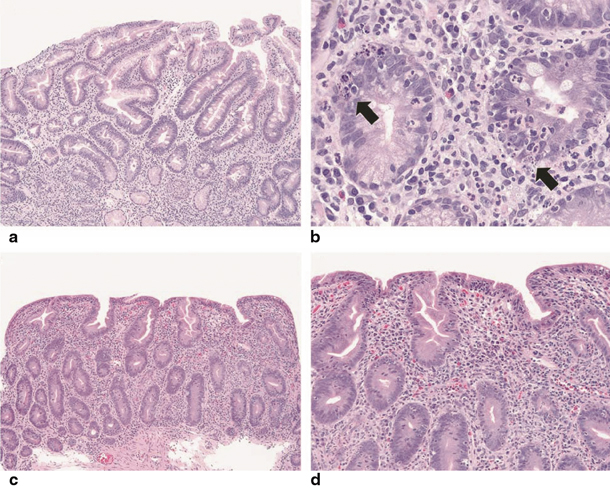
Fig. 2.1
a, b Low and high magnifications, respectively. In some cases of pediatric autoimmune enteropathy, the small intestinal biopsies show cryptitis and crypt abscesses that may obscure the salient finding of autoimmune enteropathy, crypt apoptosis (arrows). There is also an absence of Paneth cells. c, d As described in adult patients, small intestinal biopsies can demonstrate a combination of both autoimmune enteropathy and sprue-like histologic findings, characterized by severe villous blunting, marked intraepithelial lymphocytosis, diffuse mononuclear inflammatory infiltrate and prominent crypt apoptosis. Of note, goblet cells are lacking within this specimen [12]. (Reprinted by permission from Macmillan Publishers Ltd, Nature Publishing Group: Ref. [57])
Table 2.2
Histological findings in autoimmune enteropathy
Histological findings in autoimmune enteropathy |
Partial or total villous blunting/atrophy and crypt hyperplasia |
Marked infiltration of mononuclear cells, including activated T lymphocytes in the lamina propria |
Apoptotic bodies and intraepithelial lymphocytes present in the crypt/gland epithelia, but relative paucity of surface lymphocytosis |
Crypt abscesses in severe autoimmune enteropathy |
Increased expression of HLA-A, -B, -C molecules in crypt enterocytes |
AIE primarily involves the small bowel with the histologic lesions being most prominent in the proximal small intestine. However, changes have been also described in the oesophagus, stomach and colon in both pediatric and adult patients supporting the hypothesis for a diffuse disease process involving the entire gastrointestinal tract.
Recent reports describe the infiltration of the squamous epithelium by lymphocytes or eosinophils in the oesophagus. Gastric biopsies can show features of chronic nonspecific gastritis with or without reactivity. Atrophic gastritis , intestinal metaplasia and glandular destruction have been also described. There may be increased apoptosis of glandular epithelium [6, 58]. The colonic morphological lesions vary from diffuse mild active colitis with inflammatory cell infiltration to severe active chronic colitis with goblet cell depletion, Paneth cell metaplasia, distortion of crypt architecture and crypt abscess formation. An increase in intraepithelial lymphocytes has also been described [21, 58].
Diagnosis
The diagnostic criteria for AIE were originally proposed by Unsworth and Walker-Smith et al. and included (a) protracted diarrhea and severe enteropathy with small intestinal villous atrophy, (b) no response to exclusion diets, (c) evidence of predisposition to autoimmune disease (presence of circulating enterocyte antibodies or associated autoimmune disease) and (d) no severe immunodeficiency [18]. A more recent adult study proposed the updated criteria and now the diagnosis is established when all of the criteria are present (Table 2.3). The disease should be considered in the differential diagnosis in all patients presenting with severe, unexplained diarrhea requiring parenteral nutritional support particularly in infants, since AIE is the most common cause of protracted diarrhea in infancy [3]. The endoscopic examination with small bowel biopsy is the cornerstone of investigations.
Diagnostic criteria for AIE by Unsworth and Walker-Smith [18] | Updated diagnostic criteria for AIE by Akram et al. [14] |
Protracted diarrhea and severe enteropathy with small intestinal villous atrophy | Chronic diarrhea (> 6 weeks) |
No response to exclusion diets | Malabsorption |
Evidence of predisposition to autoimmune disease (presence of circulating enterocyte antibodies or associated autoimmune disease) | Small bowel histology showing partial or complete villous blunting, deep crypt lymphocytosis, increased apoptotic bodies, minimal intraepithelial lymphocytosis |
No severe immunodeficiency | Exclusion of other causes of villous atrophy, including coeliac disease, refractory sprue and intestinal lymphoma |
Presence of anti-enterocyte and/or anti-goblet cell antibody supports the diagnosis and sometimes correlates with disease improvement, but is not required to make the diagnosis |
The diagnostic work up should also include information regarding the birth and family history and the time of onset of diarrhea. The disease is characterized by secretory diarrhea , nonresponsive to bowel rest. Most of the affected infants have no history of gluten ingestion at the time of presentation. Furthermore, the lack of response to a gluten-free diet points towards AIE [5, 11, 59].
Serum immunoglobulin assays show normal immunoglobulin (Ig) M and decreased IgG attributed to protein-losing enteropathy. IgA is often within normal range, but IgA deficiency associated with villous atrophy has been also reported in AIE . T and B cell function tests, the lymphocytic subsets and polymorphonuclear cell counts are generally normal. Anti-smooth muscle, anti-nuclear and anti-thyroid microsomal autoantibodies have been identified in the course of the disease [5, 8, 22, 60].
Determination of faecal inflammatory markers, like faecal calprotectin, is a simple method that is helpful in distinguishing constitutive intestinal epithelial disorders, such as microvillus atrophy and epithelial dysplasia from immune-inflammatory etiologies such as AIE and inflammatory colitis. It has been proposed that the dramatically increased levels of faecal calprotectin in neonates and infants with immune-inflammatory disorder can distinguish these disorders from constitutive epithelial disorders with 100 % specificity [61] .
Treatment
Early recognition and accurate diagnosis of AIE are mandatory to ensure the optimal treatment. The disease is characterized by life-threatening diarrhea often nonresponsive to bowel rest. TPN represents an important step in the management of AIE for nutritional support, adequate rehydration and optimal growth [11, 14, 62]. However, the pediatric patients are not always TPN dependent during the course of the disease [19]. When the gastrointestinal involvement is less severe, elemental or low-carbohydrate-containing formula is recommended to promote enteral delivery of nutrients and calories. The potential tolerance to enteral feeds and the concomitant inflammatory changes affecting the colon make small bowel transplantation not an ideal treatment option for AIE [17, 63].
Long-term immunosuppression is the mainstay of treatment for the disease. Standard immunosuppressive therapies include corticosteroids , cyclosporine, azathioprine and 6-mercaptopurine. Steroids in the form of prednisolone or budesonide are often needed to induce remission. However, the disease can be refractory to steroids or diarrhea recurs when they are tapered [64].
Since the early 1990s, pediatric patients with AIE have been successfully treated with oral cyclosporine A. Studies have shown that a relatively low drug level (50 ng/mL) led to improvement in growth, intestinal carbohydrate absorption and small bowel histology . However, a number of patients do not respond to the medication and a possible reason is the inefficient absorption of the oral compound due to the underlying chronic enteropathy [65, 66].
Tacrolimus has been used as a therapeutic treatment option with beneficial effects in a variety of autoimmune diseases, including autoimmune hepatitis , primary sclerosing cholangitis and steroid-refractory nephrotic syndrome. Its mechanism of action is similar to cyclosporine. Both drugs block the gene activation for cytokine production by inhibiting the antigenic response of helper T lymphocytes, suppressing IL and interferon-γ [17]. Bousvaros et al. first used tacrolimus as an alternative therapy for AIE and concluded that it can be efficacious if other immunosuppressive regimens fail. Clinical improvement occurred between 1 and 4 months once therapeutic levels were achieved, and serum drug levels were obtained between 5 and 15 ng/mL. Tacrolimus’ absorption is less dependent on mucosal integrity compared to cyclosporine; however, mucosal healing improves its absorption, and the dosage should be adjusted to achieve the desired blood levels of the drug. The need for long term or lifelong treatment with tacrolimus in AIE necessitates baseline and frequent monitoring in order to prevent its potential complications, including nephrotoxicity, neurotoxicity, increased predisposition to infections and lymphoproliferative disease [67].
The combination of tacrolimus and infliximab has been proven successful in controlling the inflammation in severe AIE in both pediatric and adult patients. The synergistic effect of these agents is based on the different aspects of the immune system on which they act. Emerging evidence supports that infliximab itself is a highly effective tool for achieving clinical remission and restoring small bowel villous architecture in AIE. The drug has been introduced because of its tumour necrosis factor (TNF)-α antagonistic effect since high levels of this cytokine are being produced by intestinal intraepithelial T lymphocytes of patients with AIE. The response to infliximab is usually rapid and the quality of life of the patients improves dramatically. However, it has been recognized that aggressive immunosuppressive treatment carries a potential risk of life-threatening hypersensitivity reactions and malignancies that should be also considered [17, 68, 69].
Additional immunosuppressive therapies have been used in AIE. Cyclophosphamide is an alkylating agent related to nitrogen mustard and is a potent immunosuppressive agent used in bone marrow transplantation (BMT) conditioning regimens [70]. Low dose of oral cyclophosphamide led to resolution of the intestinal symptoms in a teenage boy with total villous atrophy, selective IgA deficiency and anti-epithelial cell antibodies [71]. However, the use of cyclophosphamide in doses up to 3 mg/kg/d has not always been successful in the management of the disease [38, 72]. Remission of symptoms and improvement of intestinal histopathology was reported in an infant with severe AIE with a single course of high-dose intravenous cyclophosphamide, approximately 20 times greater than those previously used [73].
Mycophenolate mofetil (MMF) has been proposed as an alternative therapeutic option after the successful induction of remission and the improvement of the intestinal absorption and linear growth in an infant with AIE and concomitant factor V Leiden defect [74].
IPEX Syndrome
IPEX is a rare disease and represents a systemic form of AIE characterized by immune dysfunction, polyendocrinopathy, enteropathy and X-linked inheritance. The prevalence of the syndrome remains unknown. Even though the severity of symptoms is variable, the most common feature of IPEX is the involvement of pancreas and thyroid with an early onset. Glucose intolerance can be present at birth and insulin-dependent diabetes mellitus begins often during the first year of life as a result of the complete inflammatory destruction of the pancreatic islet cells prior to the intestinal symptoms [75]. Thyroiditis presents either in the form of hyperthyroidism, or most commonly as hypothyroidism requiring substitutive therapy [13, 61] .
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


