Fig. 38.1
Overview of intestinal lipid absorption: Key genetic regulators. Following digestion of dietary and biliary lipid, the component lipid species including long-chain fatty acid (FA), monoglycerides (MG), and cholesterol are transported across the brush border membrane into the apical compartment of villus enterocytes. FA and MG undergo re-esterification into triglyceride (TG) which is mediated in a series of enzymatic steps including diacylglycerol acyltransferase 1 (DGAT1) . The newly synthesized TG exists in both cytosolic and intramembranous forms within the endoplasmic reticulum (ER) membrane, where DGAT1 (boxed) also resides. The heterodimeric ER protein MTTP physically translocates neutral lipid droplets from the ER bilayer membrane to an acceptor protein, APOB. Mammalian enterocytes synthesize a truncated APOB protein referred to as APOB48. The fusion of APOB and TG droplets, mediated by MTTP, is the critical and rate limiting step in prechylomicron assembly. Following processing in the ER, the prechylomicron undergoes vesicular transport to the Golgi, mediated by fusion with COP proteins and SAR1B. Following further maturation in the Golgi, the CM particle, containing APOB48 and a cargo of TG and cholesterol ester (CE) is transported into the lymphatic compartment and delivered into the peripheral circulation
Molecular Genetic Basis for Congenital Defects in Lipid Absorption: Overview
Several rare genetic disorders of lipid absorption have been identified and their molecular basis characterized [3–5]. These include abetalipoproteinemia (ABL), which is an autosomal recessive disorder caused by mutations or deletions in the MTTP gene [3]; familial hypobetalipoproteinemia (FHBL), an autosomal codominant disorder which is typically caused by mutations in the APOB gene [4]; chylomicron retention disease (CRD, also known as Anderson’s disease), which is an autosomal recessive disorder caused by mutations in SAR1B [5]; and one family in which congenital diarrhea was linked to a splice defect in DGAT1 [6]. The cardinal feature of enterocyte lipid droplet accumulation is common to all the disorders, but the site of lipid droplet accumulation shows subtle distinctions based on the molecular defect. Defects associated with ABL and FHBL result in lipid droplet accumulation throughout the cytoplasm but typically do not generate lipid particles within the ER lumen (Fig. 38.2, top panel). Both ABL and FHBL interrupt intestinal lipid absorption at the initiation stage of pre-CM formation and thus completely attenuate CM assembly and the generation of intracellular lipoproteins, particularly large lipoprotein particles that contain neutral lipid (cholesteryl ester and triglycerides). As a result of this block in lipoprotein assembly, neutral lipid droplets accumulate in the apical cytoplasm in ABL and FHBL. These conditions are discussed in more detail below. The defect in CRD, by contrast, is focused at a later stage in CM formation and blocks maturation and exit of CM and pre-CM particles from the ER and Golgi [7, 8]. The accumulation of lipid droplets and pre-CM particles within membranous domains is schematically illustrated in Fig. 38.2, bottom panel. CRD is discussed in a later section.
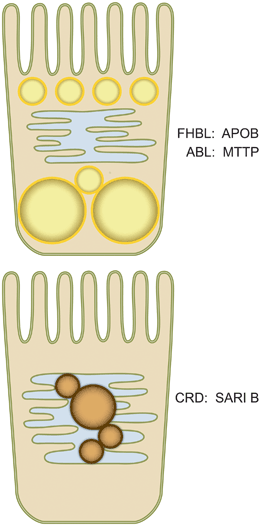
Fig. 38.2
Lipid droplet accumulation in genetic disorders of intestinal lipid absorption. Based upon the pathways outlined in Fig. 38.1, defects or mutations in APOB (responsible for familial hypobetalipoproteinemia, FHBL) or in MTTP (responsible for abetalipoproteinemia, ABL) eliminate the earliest steps in chylomicron assembly and thus lead to lipid droplet accumulation with no lipid particles visible within ER profiles (Top panel). By contrast, defects in SAR1B (responsible for chylomicron retention disease, CRD), interrupt the later stages of chylomicron maturation and processing and lead to prechylomicron accumulation (brown) within the ER lumen (Bottom panel). APOB apolipoprotein B, MTTP microsomal triglyceride transfer protein SAR1B SAR1-ADP ribosylation factor
Congenital Disorders of CM Assembly: Genetics and Clinical Features of ABL and FHBL
Defects in either of the gatekeeper genes that promote CM assembly (i.e., MTTP and APOB, respectively) each produce congenital lipid malabsorption , which generally presents in infancy as failure to thrive with characteristically low serum lipid levels and other complications (including ataxia and retinitis pigmentosa) that reflect the coincident malabsorption of fat-soluble vitamins [8]. However, the clinical phenotypes in both ABL and homozygous FHBL may be quite variable as discussed below.
Abetalipoproteinemia (ABL, OMIM #200100): Molecular Genetics and Prevalence
ABL is a rare, autosomal recessive disorder with an estimated prevalence in North America of < 1 per 100,000 [9] and for which worldwide there were about 50 cases with defined MTTP mutations reported as of 2012 [10–15]. There is strong evidence for a founder effect in Ashkenazi Jewish patients, where a conserved haplotype with a common truncating mutation (p.G865X) was discovered in Israel with a carrier frequency of 1:131 and an estimated ABL incidence of 1:69,000 [16]. Some but not all of the reported ABL probands are the offspring of consanguineous marriage, an observation replicated across different national registries [12, 16]. While a range of mutations and deletions have been described in the MTTP gene in association with ABL, affected subjects may be either homozygous for a single mutation or compound heterozygous for different mutations and there appears to be no relationship between the site and nature of these mutations and the clinical phenotypes observed [17].
Abetalipoproteinemia (ABL, OMIM #200100): Clinical Features and Management
Although typically ABL presents in infancy with failure to thrive, there is a wide age range of presentation (from infancy to > 20 years) suggesting that there are either environmental or other genetic modifiers that influence some of the clinical features. Infants typically present with diarrhea and malabsorption and the hallmark features including extremely low plasma cholesterol and TG levels, very low or undetectable levels of APOB, low to absent levels of fat-soluble vitamins and red blood cell acanthacytosis [9, 11].
The clinical phenotypes of low to undetectable levels of cholesterol and TG and APOB reflect the virtual complete block in lipoprotein secretion from both enterocytes and hepatocytes , which reflects the inability to lipidate the nascent APOB protein as it traverses the ER membrane (Fig. 38.1). The very low levels of fat-soluble vitamins (particularly vitamins A and E) reflect fat malabsorption and defective CM production. Among the neurological sequelae of unrecognized ABL is retinal degeneration, which may be progressive and result in visual impairment and which is likely a consequence of continued fat-soluble vitamin A and E deficiency [9]. The observation that ABL subjects treated upon diagnosis with long-term high-dose supplementation with vitamins A (10–15,000 IU/day) and E (100 mg/kg/day, equivalent to 150 IU/kg) [18], maintain retinal function and neurological function provides indirect but strong evidence that deficiency of these vitamins is responsible for the phenotypic outcomes, although the exact mechanisms linking ABL to neurologic deficits are yet to be fully elucidated [19]. Importantly, there was no evidence for increased oxidative stress in the plasma of ABL patients receiving long-term vitamin A and E supplementation [19]. For clinical monitoring purposes, it is worth noting that even with high-dose vitamin E supplementation, plasma levels rarely return to the normal range. This is because of the dual defect that exists in vitamin E metabolism, reflecting a block in intestinal absorption of vitamin E and also a defect in hepatic alpha tocopherol transfer to circulating lipoproteins [20]. Because circulating plasma lipid levels are so low in ABL subjects, plasma levels of alpha and gamma tocopherol may not reflect tissue (particularly adipose) concentrations, but nevertheless may be a useful surrogate to monitor long-term compliance [9].
ABL subjects require lifelong maintenance on a low-fat diet (to minimize steatorrhea) and supplementation with fat-soluble vitamins as noted above and in addition supplementation with 1000 IU/day vitamin D (which is available in a water-soluble form). Longer term, ABL subjects should be monitored for development of steatohepatitis (transaminases, liver magnetic resonance spectroscopy) which has been associated with progressive liver disease requiring liver transplantation [21] .
FHBL, OMIM #107730: Molecular Genetics and Prevalence
FHBL is an autosomal codominant disorder whose molecular basis (where known) most commonly resides in mutations in the APOB gene on chromosome 2 [22]. Unlike ABL, heterozygous FHBL subjects exhibit low circulating levels of plasma cholesterol, triglyceride, and APOB and are generally discovered in routine plasma lipid level screening tests. Heterozygous FHBL, recognized through population-based plasma lipid screening for subjects at or below the fifth percentile, is present in ~ 1:500–1:1000 [22, 23]. Homozygous FHBL by contrast is extremely rare, with perhaps 20 index cases whose molecular genetic basis is known [24]. FHBL associated with structural defects in the APOB gene represent the majority of known cases [22, 23]. The known mutations generally produce truncated forms of APOB (i.e., smaller proteins than APOB100) which are defective in undergoing lipidation despite adequate levels of MTTP (Fig. 38.1). These truncated forms of APOB may be detected using denaturing gel electrophoresis and are present at very low levels in plasma, and notably even in heterozygous subjects are found at lower abundance than those encoded by the wild-type allele. This observation implies that transcription or translation of the mutant allele in FHBL is also defective [25]. In addition, FHBL is genetically heterogeneous with several kindreds described in whom the trait is inherited in an autosomal dominant manner without mutations in the APOB gene but rather with linkage to a locus on chromosome 3p21, between D3S2407 and D3S1767 [25]. The molecular basis for the defects in FHBL subjects linking to chromosome three remains unknown.
FHBL, OMIM #107730: Clinical Features and Management
The clinical manifestations of homozygous FHBL are heterogeneous with severely affected individuals presenting in infancy or early childhood with failure to thrive and steatorrhea, while yet other affected homozygous individuals may present in adulthood with incidentally discovered hepatic steatosis and very low levels (< fifth percentile) of plasma lipids and undetectable levels of APOB100 [24]. In general, the clinical manifestations and management of severe homozygous FHBL subjects follows the same principles as alluded to above for ABL subjects, including acanthocytosis, very low levels of plasma lipids and fat-soluble vitamins, and neurologic deficits with macular degeneration and steatorrhea [26]. A more sinister concern longer term for FHBL subjects is the development of cirrhosis [27, 28] and hepatocellular carcinoma [24, 29]. The observations in relation to hepatic lipid accumulation in subjects with FHBL due to APOB truncations were not replicated in homozygous FHBL subjects linked to chromosome 3p21, reinforcing the concept that the clinical phenotypes among FHBL subjects is very variable [30, 31]. Homozygous FHBL subjects should therefore undergo regular evaluation of fat-soluble vitamin sufficiency (as outlined above) and also periodic monitoring for progressive hepatic steatosis and steatohepatitis (transaminases, liver magnetic resonance spectroscopy), again as alluded to above for ABL subjects.
CRD, OMIM #246700: Molecular Genetics and Prevalence
CRD was identified by Roy and colleagues in 1987 [32]. Although this disorder appears to be the same as that described by Anderson in 1961 [33], the term “CRD” is preferred nowadays, as it is more indicative of the pathology. CRD is a very rare, autosomal recessive disorder with fewer than 50 cases described worldwide [7], and whose molecular genetic defect resides in the SAR1-ADP ribosylation factor , type B (SAR1B), which belongs to the Ras superfamily of guanosine triphosphatase (GTPases). SAR1B mutations induce an accumulation of pre-CM transport vesicles [7], as seen on intestinal biopsies and illustrated schematically in Fig. 38.2.
SAR1B is a single polypeptide of 198 amino acids and functions as molecular switch controlled by the exchange of guanosine diphosphate (GDP) and GTP. SAR1 is cytosolic in its GDP form and is recruited to the ER by the exchange of GDP for GTP to initiate COPII-coated vesicle formation. SAR1-GTP forms a coating protein complex (COPII) with two heterodimers SEC23/24 and SEC13/31. COPII initiates budding and captures cargo to eject vesicles from the ER to the Golgi apparatus [34]. The SAR1B GTPase cycle then allows cytosolic COPII proteins to exchange on and off the membrane at specific sites on the ER to regulate cargo exit. As a result, genetic defects in SAR1B affect the transport of pre-CM cargo from the ER to the Golgi apparatus where they usually undergo additional glycosylation before being released from the enterocyte. CMs are transported from the ER to Golgi, probably inside the pre-CM transport vesicle or PCTV [35]. PCTV share several proteins including COPII (SEC13–31, SEC24 and SAR1) [36]. Siddiqi et al. [37] earlier showed that PCTV formation can occur in the absence of SAR1 and later showed that PCTV fusion with Golgi requires the presence of SAR1 [38].
The first investigation of genetic defects in eight families disclosed three frameshift (75–76 delTG, 555–558 dupTTAC, 349–1 G > C) and five missense mutations (109 G > A, 409 G > A, 537 T > A, 536 G > T, 542 T > C) [5]. The second exploration of the genetic abnormalities from eight families identified three new molecular aberrations: a stop codon mutation (364 G > T), a 5946 bp deletion (total exon 2) and a missense mutation (554 G > T) [39]. Thereafter, a new mutation G19T (changing a glutamic acid to a STOP codon) was found in the seventh exon [40] while two additional mutations were detected in exon 2 (G11D) and exon 4 (D75G) [41]. Recently, a novel homozygous deletion at position 142 (c.142delG) led to the replacement of the aspartic acid at position 48 by a threonine and gave rise to a frameshift-derived premature stop codon 17 amino acids further on (p.Asp48ThrfsX17), thereby resulting in a truncated protein (only 32 % of the length of the normal protein and 24 % of the normal sequence) [7]. Finally, the analysis of SAR1B gene mutations in Tunisian children revealed a proband homozygous for a novel nucleotide transition in exon 4 (c.184G > A), resulting in a nonconservative amino acid substitution (p.Glu62Lys) [42]. In order to provide a unifying explanation for the functional impairment of SAR1B mutated proteins, investigators have turned to information gleaned from the SAR1B crystal structure, computational analysis, and sequence alignment [43]. For example, the nonsense mutations and whole deletion of exon 2 in SAR1B yields truncated proteins that are predicted to modify the affinity of SAR1B for GDP and GTP, thereby affecting its interaction with the endoplasmic ER and other components of COPII machinery. In addition, the missense mutations in SAR1B likely yield nonfunctional proteins that alter the conformational and/or structural properties of SAR1B. Because SAR1B expression is detected in multiple different tissues, there are frequent clinical manifestations of CRD in organs beyond the gastrointestinal tract. As an example, cardiac abnormalities, myolysis, increased CK levels and cardiac abnormalities were associated with the G19T mutation [40].
Genotype–Phenotype Associations in CRD
Recent studies compared the patients of two cohorts of CRD patients from France and Canada in order to compare the severity and long-term evolution as a function of the SAR1B gene mutations [44]. In both series, most of the CRD patients were symptomatic at diagnosis (before the age of 1) and the disease was definitely more severe in infants. This is consistent with the observation that the CRD had profound impact on weight Z scores in Canadian children (1.5 ± 1 years) who were significantly younger at diagnosis than the French subjects (6.4 ± 1.3 years). The catch-up growth and bone mineral density improvement were not as encouraging in the French cohort compared to the Canadian patients, possibly because of the delayed initiation of a low-fat diet with a high polyunsaturated/saturated fatty acid ratio and of supplements of fat-soluble vitamins A, D, and E. In general, the neuro-ophthalmic manifestations detected at the time of diagnosis proved largely reversible with treatment. Although the effect of missense mutations on the protein was considered less deleterious than the those mutations that introduce stop codons or that specified deletions, the clinical phenotype (i.e., growth, neurological impairment, hepatic steatosis, lipid disturbances) was not significantly different in CRD patients based on the underlying genetic mutation [44]. According to the findings of this largest group of CRD patients so far reported, genotype–phenotype correlations are not obvious, suggesting that CRD might represent a more complex trait rather than a simple autosomal recessive disorder [44]. Additional work is required to determine whether modifier genes are involved in the ER-to-Golgi transport.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


