The Road to Transplantation Tolerance
Alan D. Salama*
Mohamed H. Sayegh†
*Imperial College London, Hammersmith Hospital, Ducane Road, London W120NN, UK; and †Transplantation Research Center, Brigham and Women’s Hospital & Children’s Hospital Boston, Harvard Medical School, Boston, Massachusetts 02115.
INTRODUCTION
Transplantation remains the treatment of choice for end organ failure of many systems, but is flawed because of the adverse reactions it promotes and its limited durability. Ideally, an organ transplant would not elicit a response by the recipient’s immune system, and would function for the duration of the recipient’s lifespan. The recipient would therefore not be subjected to the adverse effects of our current immunosuppressive agents, with the associated increased morbidity and mortality they carry and would be capable of mounting a normal immune response to invading pathogens and malignant cells (preventing clinical infectious episodes and the development of tumors). The donor organ would be treated like all the other recipient tissues, as “self,” and this state of tolerance would be robust and permanent. No intercurrent infection or inflammatory reaction would perturb this state, mirroring what is found for the majority of healthy individuals who are tolerant of their own tissues and organs. Such a state of tolerance or nonresponsiveness toward the graft in a fully immunocompetent host remains an ideal sought by all transplant immunobiologists. In this chapter we will discuss how such a state may be achieved theoretically and how near we are to reaching it, and speculate on what the future directions might be with regard to the potential agents that may be utilized to reach this goal.
MECHANISMS OF TOLERANCE
Ehrlich and Morgenroth (1) were the first to define the concept of self-tolerance, when they demonstrated that immunization with red blood cells led to antibody production in experimental animals. They found that antibodies capable of hemolyzing erythrocytes were only produced when the immunizing erythrocytes were from allogeneic or xenogeneic animals, and not when they were the recipients’ own. They went on to suggest that prevention of self-reactivity was due to absence of certain antigens on autologous erythrocytes or the production of other, less well-defined, regulatory factors (1,2). Since these early experiments were carried out, our concept of how the immune system regulates itself and achieves tolerance to self-antigens has considerably advanced. Landmark studies by Owen (3) and Billingham, Brent, and Medewar (4) provided evidence that exposure to antigens during the neonatal period allowed for the development of tolerance toward those antigens regardless of their origin.
During development of the immune system, a huge amount of effort and energy is expended in programming the system not to react to self-antigens. Natural tolerance is achieved through two main processes; the first, termed central or clonal selection, involves the elimination of cells directed against self-determinants and the positive selection of
those that are not. The second process, termed peripheral tolerance, utilizes numerous different mechanisms, including functional inactivity of autoreactive cells (anergy); suppression of autoreactive cells by other cells or factors (regulation); indifference to the stimulating antigen (ignorance); and in certain cases prevention of autoreactive cells and their targets from interacting (sequestration) (5,6) (See Fig. 11.1). The main cellular components coordinating the cognate immune response, and hence determining self-tolerance, are the T and B lymphocytes. However, since the T cells are the main orchestrators of the immune response, providing essential help to antigen-specific B cells and cytotoxic T cells, as well as specificity and help in the delayed type hypersensitivity response, they act as gatekeepers of immune reactivity and nonreactivity. Mechanisms of tolerance induction operate during development in the thymus and bone marrow but also once the mature cells have been generated and are found in the periphery. The precise requirements for tolerance differ in the T- and B-cell compartments (7,8), with T-cell tolerance being achieved more easily, with lower antigen doses and persisting for longer than B-cell tolerance (6). Thus, teleologically, it may be more important to regulate helper T-cell responses rather than those of B cells, since the latter are reliant on T-cell help in the majority of cases. Our understanding of the mechanisms promoting tolerance comes from the investigation of both auto- and alloimmunity, hence the discussion that follows interweaves through both systems.
those that are not. The second process, termed peripheral tolerance, utilizes numerous different mechanisms, including functional inactivity of autoreactive cells (anergy); suppression of autoreactive cells by other cells or factors (regulation); indifference to the stimulating antigen (ignorance); and in certain cases prevention of autoreactive cells and their targets from interacting (sequestration) (5,6) (See Fig. 11.1). The main cellular components coordinating the cognate immune response, and hence determining self-tolerance, are the T and B lymphocytes. However, since the T cells are the main orchestrators of the immune response, providing essential help to antigen-specific B cells and cytotoxic T cells, as well as specificity and help in the delayed type hypersensitivity response, they act as gatekeepers of immune reactivity and nonreactivity. Mechanisms of tolerance induction operate during development in the thymus and bone marrow but also once the mature cells have been generated and are found in the periphery. The precise requirements for tolerance differ in the T- and B-cell compartments (7,8), with T-cell tolerance being achieved more easily, with lower antigen doses and persisting for longer than B-cell tolerance (6). Thus, teleologically, it may be more important to regulate helper T-cell responses rather than those of B cells, since the latter are reliant on T-cell help in the majority of cases. Our understanding of the mechanisms promoting tolerance comes from the investigation of both auto- and alloimmunity, hence the discussion that follows interweaves through both systems.
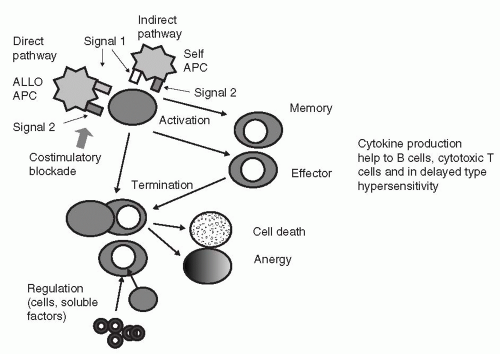 FIG. 11.1. The fate of alloreactive T cells. Upon antigen engagement and in the presence of adequate costimulatory signals, the alloreactive T cell becomes activated, proliferates, and can subsequently undergo a number of different fates. The first is differentiation to an effector cell, orchestrating the immune response directed toward the target antigen. Some of the T cells will differentiate into memory cells, able to provide rapid recall responses upon antigenic restimulation. Other cells will have their effector functions terminated by dying (through a process of apoptosis by passive- or activation-induced cell death, PCD and AICD, respectively), becoming regulated by other cells or soluble factors, or in the absence of adequate costimulation, becoming anergic, a state of nonresponsiveness to further antigen stimulation. The majority of alloreactive T cells will proliferate, produce cytokines, and provide help to other limbs of the immune response, then die through AICD. (Adapted from Salama AD, Remuzzi G, Harmon WE, et al. Challenges to achieving clinical transplantation tolerance. J Clin Invest 2001;108:943-948.) |
Clonal Selection
The concept of eliminating the most autoreactive lymphocytes, resulting in a “hole” in the immune repertoire, while the cells with less autoreactive potential are retained, dates back to Burnet (9). The clonal deletion theory as it applies to T lymphocytes, states that T cells developing in the thymus which react most strongly with antigens presented by self-MHC (major histocompatibility complex) are eliminated (negative selection) while those with lesser avidity continue to develop, and are subsequently exported to the periphery (positive selection). Thus, the majority of high avidity autoreactive T cells should be eliminated during T-cell ontogeny; however, some lower avidity autoreactive T cells may escape to the periphery and are therefore kept in check by a number of different mechanisms so as not to induce autoimmune disease. Data confirming clonal deletion have been subsequently found (10,11).
T-cell selection begins after the recent thymic immigrants, arriving from hemopoietic tissues, have rearranged their T-cell receptor and upregulated both CD4 and CD8 antigens. Ultimately, only 3% of the total number of CD4+CD8+ double positive cells are exported from the thymus having developed into single positive CD4+CD8− or CD8+CD4− cells (12). In the absence of T-cell receptor (TcR) engagement with self-MHC molecules during the lifespan of double positive (CD4+CD8+) thymocytes, the cells die, while those T cells that bind with a certain affinity to polymorphic regions of self-MHC molecules survive as a
result of expression of the anti-apoptotic protein Bcl-2 (12). However, TcR engagement may result in either elimination and negative selection, or differentiation and positive selection (13). Which one of these two occurs is in part related to the particular peptide bound to the MHC molecule, its concentration, the TcR affinity for the MHC-peptide complex, and other secondary costimulatory signals provided to the T cell (14, 15, 16, 17). This allows those cells with the strongest avidity for self-MHC and antigen to be deleted (18). The cells undergo apoptosis or are rendered anergic, a state in which they are hyporesponsive following an interaction with antigen, neither proliferating nor producing interleukin IL-2. Negative selection is dependent on the presentation of antigen by dendritic cells or medullary thymic epithelial cells (19) to developing T cells. The exact contribution each makes has been debated and is partly dependent on the experimental system that has been studied (18,20). Further development of the surviving thymocytes involves upregulation of a number of surface molecules including the TcR-CD3 complex, CD5, and CD69, and the loss of either CD4 or CD8 (13). Thus, the cells emigrating from the thymus are of low to intermediate affinity for self-MHC and antigen, and are positive for either CD4 or CD8.
result of expression of the anti-apoptotic protein Bcl-2 (12). However, TcR engagement may result in either elimination and negative selection, or differentiation and positive selection (13). Which one of these two occurs is in part related to the particular peptide bound to the MHC molecule, its concentration, the TcR affinity for the MHC-peptide complex, and other secondary costimulatory signals provided to the T cell (14, 15, 16, 17). This allows those cells with the strongest avidity for self-MHC and antigen to be deleted (18). The cells undergo apoptosis or are rendered anergic, a state in which they are hyporesponsive following an interaction with antigen, neither proliferating nor producing interleukin IL-2. Negative selection is dependent on the presentation of antigen by dendritic cells or medullary thymic epithelial cells (19) to developing T cells. The exact contribution each makes has been debated and is partly dependent on the experimental system that has been studied (18,20). Further development of the surviving thymocytes involves upregulation of a number of surface molecules including the TcR-CD3 complex, CD5, and CD69, and the loss of either CD4 or CD8 (13). Thus, the cells emigrating from the thymus are of low to intermediate affinity for self-MHC and antigen, and are positive for either CD4 or CD8.
For clonal deletion within the thymus to occur, the self-antigen has to be presented on the surface of antigen-presenting cells (APC). As such, the antigen has to be expressed in the thymus or reach it in adequate concentration via the circulation. However, this process cannot account for tolerance to antigens that are generated after the T-cell repertoire has developed, or which are expressed in a tissuerestricted manner, or induced only under certain circumstances. For such antigens it was assumed that peripheral mechanisms of tolerance were sufficient to keep T-cell reactivity in check. However, recent data have suggested that the number of autoantigens expressed in the thymus is significantly greater than was previously appreciated. A number of self-antigens, which are targets of autoimmune processes and were previously thought to be limited in their tissue distribution, have been found to be expressed, at least at the level of RNA, in human thymic tissue (21, 22, 23). The implication of these data is that a larger number of self-proteins are synthesized in the thymus than hitherto thought, including some sequestered (such as retinal) and tissue-specific (such as insulin) antigens. The importance of thymic expression of self-antigens has been shown in a transgenic animal model of autoimmune gastritis, where thymic expression of the autoantigen was sufficient to prevent the onset of autoimmunity (24). Also, levels of thymic expression of certain autoantigens have been shown to inversely correlate with the predisposition to develop autoimmune disease (16,25). In human disease, levels of expression of insulin within the thymus appear to correlate with the allele of the diabetes-susceptibility locus, such that higher levels of thymic insulin expression are associated with alleles protective against diabetes (26,27). Furthermore, the duration of autoantigen expression within the thymus appears to determine the stringency of tolerance to the antigen (28,29). Not surprisingly, attempts have been made to induce tolerance through intrathymic antigen injection. Intrathymic inoculation of both auto- and alloantigens has been reported to induce tolerance, with T-cell tolerance being easier to achieve than B-cell, and Th1 responses easier than Th2 (7,8,30), although this has not been a universal finding when different models and protocols have been followed (31,32).
Thus, the resistance to developing autoimmunity (and the induction of transplant tolerance under certain circumstances, such as following bone marrow transplantation) may in part be related to the capacity to delete auto- (or allo) reactive T cells within the thymus (18,33), and this may be related to the level and kinetics of thymic antigen expression. This is consistent with the findings that different antigen doses may induce negative or positive selection in thymic organ cultures, with low levels determining positive selection whereas higher levels appear to be associated with negative selection and tolerance. This has been termed the affinity-avidity model of thymic selection (17).
Despite negative selection, autoreactive T and B cells do escape into the periphery and such cells can be found in normal individuals (34,35). However, autoimmune disease is uncommon. These cells are prevented from initiating damage by a number of different regulatory mechanisms, each of which contributes toward tolerance to a variable degree depending on the experimental system studied. Data on such mechanisms in human disease remain sparse.
The Role of Apoptosis
One fate of activated T cells is programmed cell death termed activation-induced cell death (AICD). This is mediated by the interaction of Fas (CD95) with its ligand (Fas-L, or CD95L) on T cells, and can occur in developing thymocytes as well as mature T cells (36). IL-2 acting on its receptor and activating the STAT5 signaling pathway potentiates the upregulation of Fas-L and downregulates Bcl2 expression on T cells, thus sensitizing them to AICD (37,38). Inadequate AICD may therefore play a role in tolerance breakdown and autoimmunity. Conversely, augmented AICD can promote tolerance through elimination of allo- or autoreactive lymphocytes, and this underpins certain tolerogenic protocols and therapies (39, 40, 41). Central to this process is IL-15 acting as a growth and survival factor for T cells, and IL-2 sensitizing them through the STAT5 pathway to AICD (37,38,42).
Fas-mediated apoptosis may also be responsible for elimination of B cells, since Fas is upregulated on activated B cells and Fas-deficient mice are characterized by a number of different autoantibodies. Prolonged survival of autoreactive B cells may therefore contribute to autoimmune disease (43).
Anergy
Successful T-cell activation requires the presentation of peptide on the appropriate MHC molecule to the TcR, termed “signal 1” and costimulatory signals termed “signal 2” (44),
the most significant of which are the B7-CD28 and CD40-CD154 interactions (45). T cells stimulated by low or high antigen doses or in a costimulation deficient manner may be rendered anergic that is hyporesponsive to further antigenic stimulation. In some models these cells are capable of certain effector functions, such as production of interferon-γ, but cannot produce IL-2 or undergo autocrine proliferation. However, this state can be overcome by the addition of exogenous IL-2 (45). This phenomenon was first systematically investigated by Dresser (46) using a system of antibody production following injection with bovine gamma-globulin (BGG). Immunization of animals with large quantities of soluble BGG rendered the animals unresponsive to further antigenic challenge with BGG. A dose-response effect was seen with increasing dosage leading to greater degrees of unresponsiveness. This was the case whether BGG rechallenge was with or without Freund’s complete adjuvant. However, if Freund’s complete adjuvant was coadministered with the initial immunization, the unresponsiveness to subsequent challenge was abrogated and immunity developed (46). The interpretation of these experiments is that anergy is induced following the presentation of antigen by costimulation-deficient APC, as a lack of anergy and induction of immunity was found if the experiments were carried out with the addition of Freund’s adjuvant containing mycobacterial antigens, known to upregulate costimulatory molecules (47). Similar findings have been reported with T-cell responsiveness. Anergic T cells formed in vivo following the intravenous injection of high-dose antigen were found to express similar levels of IL-2 receptor α chain (CD25), CD4, and TcR as did responsive T cells, but did not produce IL-2, nor did they proliferate or synthesize much DNA in response to antigen stimulation (48). IL-2 is a major T-cell growth factor, acting in an autocrine and paracrine manner on activated T cells. Furthermore, IL-2 production by activated T cells is dependent on the cells receiving signal 2 through the B7-CD28 system, acting on the levels of IL-2 transcription and the stability of IL-2 mRNA (45). IL-2 is therefore a key player in regulating T-cell responsiveness and tolerance (see AICD above) and is part of a complex interactive pathway with other cytokines such as IL-15 (49). The importance of the APC in dictating the outcome of an immune response has been confirmed by other studies. Immunity results from presentation by professional APC, carrying the full complement of costimulatory molecules, whereas tolerance is achieved using nonprofessional APC, lacking such molecules (50). However, other data have suggested that, in vivo at least, anergy requires the presence of some B7 costimulatory molecules, as following utilization of B7 antagonists, a state of immunological ignorance is induced. Anergy in this model is dependent on the interaction of B7 and CTLA4, while the interaction of B7 and CD28 results in immunity (51). It is possible that unresponsive states may be achieved by a number of different routes, some dependent on B7 and some requiring a lack of costimulation. Alternatively, it may be that low levels of B7 encourage CTLA4 signaling and higher levels, induced by adjuvants or nearby inflammation, increase signaling through CD28. This is reminiscent of the “danger model” of autoimmunity and tolerance (52). Furthermore, a role for T-cell-expressed B7 molecules in the ligation of CTLA4 and the induction of unresponsiveness has recently been proposed (53). This appears to occur only when costimulation by the APC is either absent or limiting.
the most significant of which are the B7-CD28 and CD40-CD154 interactions (45). T cells stimulated by low or high antigen doses or in a costimulation deficient manner may be rendered anergic that is hyporesponsive to further antigenic stimulation. In some models these cells are capable of certain effector functions, such as production of interferon-γ, but cannot produce IL-2 or undergo autocrine proliferation. However, this state can be overcome by the addition of exogenous IL-2 (45). This phenomenon was first systematically investigated by Dresser (46) using a system of antibody production following injection with bovine gamma-globulin (BGG). Immunization of animals with large quantities of soluble BGG rendered the animals unresponsive to further antigenic challenge with BGG. A dose-response effect was seen with increasing dosage leading to greater degrees of unresponsiveness. This was the case whether BGG rechallenge was with or without Freund’s complete adjuvant. However, if Freund’s complete adjuvant was coadministered with the initial immunization, the unresponsiveness to subsequent challenge was abrogated and immunity developed (46). The interpretation of these experiments is that anergy is induced following the presentation of antigen by costimulation-deficient APC, as a lack of anergy and induction of immunity was found if the experiments were carried out with the addition of Freund’s adjuvant containing mycobacterial antigens, known to upregulate costimulatory molecules (47). Similar findings have been reported with T-cell responsiveness. Anergic T cells formed in vivo following the intravenous injection of high-dose antigen were found to express similar levels of IL-2 receptor α chain (CD25), CD4, and TcR as did responsive T cells, but did not produce IL-2, nor did they proliferate or synthesize much DNA in response to antigen stimulation (48). IL-2 is a major T-cell growth factor, acting in an autocrine and paracrine manner on activated T cells. Furthermore, IL-2 production by activated T cells is dependent on the cells receiving signal 2 through the B7-CD28 system, acting on the levels of IL-2 transcription and the stability of IL-2 mRNA (45). IL-2 is therefore a key player in regulating T-cell responsiveness and tolerance (see AICD above) and is part of a complex interactive pathway with other cytokines such as IL-15 (49). The importance of the APC in dictating the outcome of an immune response has been confirmed by other studies. Immunity results from presentation by professional APC, carrying the full complement of costimulatory molecules, whereas tolerance is achieved using nonprofessional APC, lacking such molecules (50). However, other data have suggested that, in vivo at least, anergy requires the presence of some B7 costimulatory molecules, as following utilization of B7 antagonists, a state of immunological ignorance is induced. Anergy in this model is dependent on the interaction of B7 and CTLA4, while the interaction of B7 and CD28 results in immunity (51). It is possible that unresponsive states may be achieved by a number of different routes, some dependent on B7 and some requiring a lack of costimulation. Alternatively, it may be that low levels of B7 encourage CTLA4 signaling and higher levels, induced by adjuvants or nearby inflammation, increase signaling through CD28. This is reminiscent of the “danger model” of autoimmunity and tolerance (52). Furthermore, a role for T-cell-expressed B7 molecules in the ligation of CTLA4 and the induction of unresponsiveness has recently been proposed (53). This appears to occur only when costimulation by the APC is either absent or limiting.
It has been directly demonstrated that anergic T cells persist in vivo with defective proliferative responses, IL-2 and TNF-α production, but that these defects were reversible with time. They persisted for a number of weeks or while antigen was still administered. However, the anergic state was not overcome by supplementation with T-cell growth factors, derived from activated bystander cells (54). Thus peripheral tolerance would be maintained so long as the antigen is still present, and available to the T cells despite other ongoing immune responses.
The situation is somewhat complicated by the recent recognition of a myriad of other costimulatory molecules, from both the B7 and TNF families (55,56) which can induce T-cell activation in the absence of B7 signaling. Although initially thought of as representing multiple redundant pathways, it has recently been appreciated that some molecules appear to act on particular cellular compartments and at particular time points (55). Furthermore, it is apparent that memory T cells, CD8+ T cells, and NK cells rely less on B7-CD28 interactions in order to achieve full activation (see below). The exact role that these alternative pathways play in inducing T-cell anergy remains to be clarified.
Anergic cells may themselves act to attenuate the responsiveness of other T cells, by downregulating the costimulation molecules on dendritic cells and thus inhibiting effective antigen presentation (57). This appears to be dependent on cell: cell contact mechanisms, and represents one end of the spectrum of regulatory cells (see below).
Immunological Ignorance
In some cases tolerance is maintained to self-antigens without deletion or anergy of the autoreactive T cells, rather they ignore the (auto)-antigen under normal circumstances and are not activated. However, following activation they recognize their antigenic epitope and mediate tissue damage. This has been demonstrated in transgenic mice expressing lymphocytic choriomeningitis virus (LCMV) glycoprotein in the pancreatic β cells crossed with mice transgenic for a TcR against the LCMV glycoprotein (58). T cells responsive to the antigen can be isolated from the animals, confirming that clonal deletion has not taken place. However, neither insulitis nor diabetes was found until the animals were infected with LCMV. The specificity of the response was shown by the lack of disease when mutant strains of LCMV were used, which did not possess the appropriate LCMVglycoprotein epitope. Ignorance in this case may be due to the inability of the pancreatic cells to activate the T cells,
probably as a result of inadequate second signals, although why anergy was not induced is unclear. Furthermore, these data appear to be at odds with the findings of cross-presentation of tissue antigens, by APC, to T cells in draining lymph nodes. For example, ovalbumin (OVA) expressed in the pancreas or kidney can be presented to OVA-specific T cells in the draining lymph nodes, resulting in T- cell activation and proliferation (59). This may cause deletion of the responding T cells following activation by AICD and lead to tolerance (60). These two apparently contradictory findings may be explained by differences in levels of tissue antigen expression, since lower levels of OVA expression can lead to ignorance (61). However, these low levels must be sufficient for T-cell recognition to occur, in order to explain the findings of T-cell-mediated injury following T-cell activation in the LCMV model.
probably as a result of inadequate second signals, although why anergy was not induced is unclear. Furthermore, these data appear to be at odds with the findings of cross-presentation of tissue antigens, by APC, to T cells in draining lymph nodes. For example, ovalbumin (OVA) expressed in the pancreas or kidney can be presented to OVA-specific T cells in the draining lymph nodes, resulting in T- cell activation and proliferation (59). This may cause deletion of the responding T cells following activation by AICD and lead to tolerance (60). These two apparently contradictory findings may be explained by differences in levels of tissue antigen expression, since lower levels of OVA expression can lead to ignorance (61). However, these low levels must be sufficient for T-cell recognition to occur, in order to explain the findings of T-cell-mediated injury following T-cell activation in the LCMV model.
Another example of immunological ignorance is the lack of reactivity of cytotoxic T cells (CTL) to subdominant peptide antigens presented by a lymphoma tumor cell line, which is constitutively negative for the costimulatory molecule B7.1. These CTL were not anergized, since they could respond to tumor cells transfected with B7.1, but were not active against particular T-cell epitopes unless B7.1 was present on the tumor cell. A possible mechanism underlying this lack of reactivity is again related to levels of antigen expression. It may be that subdominant peptides are expressed on class I molecules at levels that are insufficient to normally activate the CTL. However, in the presence of costimulation, the activation threshold is reduced, allowing them to become activated by the subdominant epitopes (62). It remains unclear whether levels of alloantigen expression would be low enough to promote ignorance as a physiological mechanism of nonresponsiveness.
Cytokine Responses
It is now well established that T-cell effector responses in rodents and humans can be separated according to the cytokine profile that the T cells secrete (63). Th1 cells produce IFN-γ, IL-2, lymphotoxin, and TNF-α, and are involved in cell-mediated responses and some antibody-mediated responses, whereas Th2 cells produce IL-4, IL-5, IL-10, and IL-13 and provide help for B cells to produce IgM, IgA, and particularly IgE antibodies. Differentiation along the Th1 pathway is under the influence of IL-12, IFN-γ, and IFN-α, whereas IL-4 appears to be the essential Th2 pathway differentiation cytokine. Regulation of these responses is tightly controlled by the transcription factors, T-bet and GATA3 (64, 65, 66). Th1 cytokines antagonize some of the effects of Th2 cytokines and vice versa. This T-helper-cell differentiation is not as clear-cut in human subjects as it is in animals, and the relative balance between Th1 and Th2 responses is probably more relevant than absolute division of function. The balance between these different effector functions and the bias for one or the other may therefore have major influences on the regulation of immune reactivity and disease.
A number of models of cell-mediated autoimmune disease in mice have demonstrated polarized cytokine profiles, generally with Th1 responses being dominant in disease pathogenesis (67). Furthermore, Th2 cytokines have been shown to be protective in Th1-induced autoimmune diseases, as have interventions aimed at inhibiting the generation of Th1 cytokines. Moreover, during remission of certain Th1 diseases, the Th2 profile may be dominant (68), while adoptive transfer of Th2 cells alongside Th1 cells can attenuate acute disease and prevent relapses, demonstrating that the Th2 cells can exert regulatory effects on Th1 cells (69, 70, 71). Similarly, in alloimmune responses a Th2 cytokine deviation may confer protection from rejection in both rodents and humans (72, 73, 74). However, it should be noted that the situation is not so clear-cut, as STAT4- and STAT6-deficient animals reject their allografts with a similar tempo to wild type (75) , as do IFN-deficient recipients (although they do not develop chronic rejection) (76) (see below). Moreover, it should be noted that there is diversity and redundancy in Th2 responses with dissociated cytokine regulation reported, such that IL-4 and IL-5 may be upregulated separately (77). This means that the conclusions drawn from studies in which only some of the Th2 cytokines are analyzed need to be interpreted with caution.
The deviation of a T-cell cytokine response to a single antigen may result in infectious tolerance toward other related antigens (78) or coadministered antigens (79). Cytokine deviation may also in part underlie the tolerogenic effects of altered peptide ligands, and immunization with incomplete Freund’s adjuvant (IFA). Recognition of altered auto- or alloantigenic peptides by T cells can lead to alterations in their cytokine profile, from a Th1 to a Th2 response, with protection from disease (77,80,81).
Since many of these data are generated in rodent models, it is important to note that differences in animal strain may alter the nature of the response with respect to cytokine production and immunoglobulin subclass, but not necessarily the disease profile (82). Moreover, differences in species exist with respect to the ability of antibodies to fix complement, such that in mice only Th1-dependent IgG subclasses are complement fixing, while in rats both Th1- and Th2-dependent IgG are, explaining why Th1 responses in the mouse may be responsible for certain diseases, whereas both Th1 and Th2 responses can induce such diseases in rats. These data demonstrate that cytokine polarization and deviation may alter the nature of the response, but may have little effect on disease outcome in certain animals. Differences in cytokine responses may be due to the genetic background of particular animal strains, which produce predominantly Th1 or Th2 responses, but are also influenced by the nature of the antigen (83). Moreover, and most important, in patients a mixed Th1 and Th2 cytokine response is generally found when T cells or antibody subtypes are analyzed (84), suggesting that, unlike mice, humans do not produce such polarized responses, making cytokine deviation a less appealing therapeutic option (see below).
There is also evidence that the antigen dose and the nature of the antigen-presenting cells play a role in determining the cytokine bias and the outcome of the immune response. Low antigen doses appear to favor Th2 responses, and high doses Th1. While in allotransplantation the requirement for Th1 cytokines for tolerance induction (through activation-induced death) is only found when the T-cell clone size is large (85). One additional factor regulating the cytokine profile that is ultimately produced is costimulation through particular molecules. B7 molecules on APC interacting with CD28 on T cells are intimately involved with T-cell IL-2 production and the regulation of responsiveness to IL-12 through the upregulation of the IL-12β2 receptor subunit. They therefore play a role in deviation toward Th1 responses (86). Moreover, by antagonizing the B7-CD28 interaction, it is possible to generate predominant Th2 responses in both auto- and alloimmunity (72,80). This is in part due to the decreased ability of T cells to induce production of IL-12 by APC in the absence of CD28 signaling (86). Interestingly, while blockade of the B7-CD28 pathway can induce tolerance in Th1-predominant (STAT6-deficient) and Th2-predominant (STAT4-deficient) animals, CD40-CD154 blockade can only do so in Th2-predominant animals (75).
Whether Th2 cytokine deviation is an adequate regulatory mechanism to inhibit Th1 immune responsiveness is in part dependent on the strength and size of the immune response. Hence, in a pancreatic islet transplant model, where rejection is generally Th1 dependent, fully MHC-mismatched grafts had similar survival with or without anti-IL12 mAb treatment. However, MHC-matched but minor antigenmismatched grafts had significantly prolonged graft survival following this treatment. Furthermore, if the indirect pathway of allorecognition was the only pathway available (using MHC II-deficient mice as donors), deviation to a Th2 response by the anti-IL12 mAb again significantly improved graft outcome (87). In these latter two cases, the responding T cells were less abundant in number and antigen was presented on self-MHC, by the indirect pathway, in an analogous fashion to autoimmune disease. However, in fully MHC-mismatched grafts, there is a far greater direct pathway immune response, with greater numbers of responding T cells, which may not be overcome with cytokine deviation.
Related posts:
 Detection of Recipient Pretransplant Alloreactivity
Detection of Recipient Pretransplant Alloreactivity
 Surgical Issues in the Transplant Recipient
Pregnancy and Gender Issues in the Renal Transplant Recipient
Surgical Issues in the Transplant Recipient
Pregnancy and Gender Issues in the Renal Transplant Recipient
 Posttransplantation Liver Disease
Posttransplantation Liver Disease
 Cytomegalovirus in Renal Transplantation
Cytomegalovirus in Renal Transplantation
 Medical Complications of the Eyes, Nasopharynx, Dentition, Oropharynx, and Hearing in the Kidney Transplant Recipient
Medical Complications of the Eyes, Nasopharynx, Dentition, Oropharynx, and Hearing in the Kidney Transplant Recipient
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


