The Nonneoplastic Stomach
Embryology
The stomach develops from a fusiform foregut swelling at approximately 4 weeks’ gestation. It originates in the neck and descends into the abdomen during the next 8 weeks. The enlarging thoracic contents push the stomach caudally. The gastric curvature develops during the 6th to 7th fetal week. Simultaneously, the dorsal stomach rotates to the left. In the ninth week, a diverticulum appears in the upper stomach, which subsequently merges with and lengthens the greater curvature. The stomach rotates 90 degrees so that the greater curvature lies on the left, and the distal end becomes anchored by a short ventral mesentery, the bile duct, and the vitelline artery (1).
Gastric development is more complex than other parts of the gut due to the different epithelial types that populate different areas of the stomach. These areas constitute a complex epithelial system organized in a highly structured, continually renewing architecture. Embryonal differentiation is regulated via several signaling cascades, an important one of which starts with the transcription factor Sonic hedgehog (Shh), which binds to its receptor Patched (Ptc). The Shh signaling system helps maintain normal gastric glandular architecture (2). Shh is expressed in parietal cells and its receptor Ptc is present in chief cells (3).
The stomach is initially lined by stratified or pseudostratified epithelium; later, cuboidal cells replace it. As secretions accumulate, droplets and vacuoles coalesce to form the gastric lumen. The first differentiated cell type to appear is the mucous neck cell, which acts as a progenitor for the other cell types. Gastric pits are well developed by 5 to 7 weeks. Gastric glands begin to develop at 11 to 14 weeks (1); they grow by progressively branching, a process that continues until birth. Parietal cells appear by 9 to 11 weeks (Fig. 4.1). Endocrine cells begin to appear at the second week of fetal life; a full spectrum of endocrine cells is present by week 11. Mesoderm surrounding the stomach differentiates into the gastric connective tissue and the muscularis propria by the end of the second fetal month. The muscularis mucosae forms by the 20th week.
Gastric Physiology
The stomach exhibits major motor, secretory, digestive, hormonal, and mucosal barrier functions, some of which will be briefly summarized here.
Mucosal Barrier
One of the incredible facets of gastric physiology is that the acid-containing stomach is able to withstand the detrimental effects of its intraluminal contents. In order to do this, a complex mucosal cytoprotection system has evolved that protects the stomach without inhibiting gastric acid secretion. Mucosal defenses include pre-epithelial, epithelial, and postepithelial mechanisms (Fig. 4.2). Adherent mucus provides a stable unstirred layer that supports surface neutralization of acid by mucosal bicarbonate and acts as a permeability barrier to luminal pepsin (4). (Surface mucus is hydrophobic and water repellent.) Surface-active phospholipids are produced by mucous neck cells and parietal cells. Parietal cells pump one HCO3– ion across the basal membranes for every H+ they secrete into the canaliculi (5). HCO3– is picked up by mucosal capillaries and carried to the basal part of surface foveolar cells. The bicarbonate ions are then secreted into the overlying mucous layer, where they are trapped by glycoproteins in the mucus, increasing the pH in the unstirred layer from approximately pH 2.0 in the gastric lumen to approximately 7.0 at the mucosal surface. This creates a pH gradient that traps and neutralizes most hydrogen ions as they enter the unstirred mucous layer (6). Maintenance of the pH gradient depends on both the secretion rate of bicarbonate and the thickness of the mucous gel layer (6). Mucus also lubricates the stomach facilitating food movement along the gastric lining, without causing mucosal abrasions. Its glycoproteins play a major role in resistance to injury by maintaining the viscoelastic and permeability properties of the mucous gel. Foveolar cells secrete lipid into the mucus that coats the epithelium lining the gastric lumen with a nonwettable surface, protecting the mucosa against the action of water-soluble H+ and pepsin (7). (Pepsin can destroy the polymeric structure of this glycoprotein layer, solubilizing the surface mucous gel and liberating degraded glycoprotein subunits into the gastric lumen.)
An adequate mucosal blood flow is critical to maintaining the mucosal barrier since it brings oxygen and nutrients to the luminal surface and removes hydrogen ions from the same region (8). The autonomic nervous system, peptidergic nerves (8), nitric oxide (9), prostaglandins (10), epidermal growth factor (EGF), and transforming growth factor-alpha
(TGF-α) all regulate mucosal blood flow. Interruption of the mucosal blood flow (as occurs in stress gastritis) results in decreased intramucosal pH and ulceration. Junctional complexes, basolateral membranes, and the basal lamina are also major structural components of the gastric mucosal barrier (11). Cytoprotectants (prostaglandins, immunoglobulins, sulfhydryl donors such as glutathione, and neuropeptides) are also naturally present in the gastric mucosa (10,12). Prostaglandins aid in mucosal protection (10) by mediating mucus and bicarbonate secretion, inhibiting acid secretion, regulating mucosal blood flow, maintaining surface-active phospholipids, and mediating the protective actions of EGF and TGF-α (13). Prostaglandins also modulate the inflammatory response by inhibiting release of tumor necrosis factor (TNF) from macrophages (14) and TNF plus other inflammatory mediators from mast cells (15).
(TGF-α) all regulate mucosal blood flow. Interruption of the mucosal blood flow (as occurs in stress gastritis) results in decreased intramucosal pH and ulceration. Junctional complexes, basolateral membranes, and the basal lamina are also major structural components of the gastric mucosal barrier (11). Cytoprotectants (prostaglandins, immunoglobulins, sulfhydryl donors such as glutathione, and neuropeptides) are also naturally present in the gastric mucosa (10,12). Prostaglandins aid in mucosal protection (10) by mediating mucus and bicarbonate secretion, inhibiting acid secretion, regulating mucosal blood flow, maintaining surface-active phospholipids, and mediating the protective actions of EGF and TGF-α (13). Prostaglandins also modulate the inflammatory response by inhibiting release of tumor necrosis factor (TNF) from macrophages (14) and TNF plus other inflammatory mediators from mast cells (15).
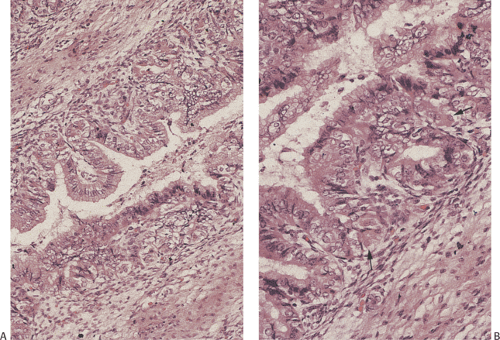 FIG. 4.1. Gastric mucosa of a 10-week-old fetus. A: Medium power showing the presence of a well-defined lumen lined by columnar epithelial cells with primitive glands. The muscularis mucosae is just beginning to form. B: Higher magnification showing the presence of parietal cells (arrows). |
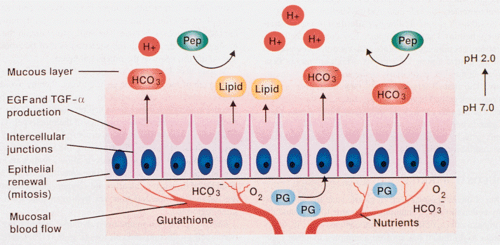 FIG. 4.2. Mucosal defenses. A mucous layer that contains a pH gradient overlies the surface epithelium. Bicarbonate ions are pumped into this layer along with lipids secreted by the lining epithelium. The epithelium is bound together by intercellular tight junctions. The epithelial cells lie on intact basement membrane and produce the epidermal growth factor (EGF) and transforming growth factor-α (TGF-α). The underlying blood supply in the lamina propria brings bicarbonate to the surface-lining cells from the parietal cells where it was produced. The mucosal blood flow also brings oxygen and nutrients. Prostaglandins (PGs) are made within the stroma. The stroma contains antioxidants such as glutathione. Pep, pepsin. |
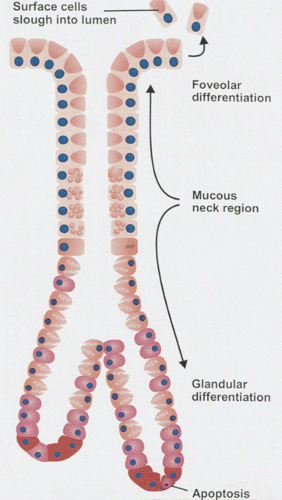 FIG. 4.3. Mucosal renewal. The mucous neck region contains stem cells and is the generative zone. From here, foveolar cells begin to differentiate and migrate toward the surface to be exfoliated. Other cells developing in this area migrate downward to form the epithelium of the oxyntic, cardiac, and antropyloric glands. These glandular cells die by apoptosis. |
Another aspect that protects the gastric mucosa is its ability to proliferate and rapidly replace damaged surface epithelial cells. The gastric epithelium maintains a dynamic equilibrium between cell production and cell loss (Figs. 4.3 and 4.4) (16). The surface epithelium is renewed every 4 to 8 days. Gastrointestinal and nongastrointestinal hormones, growth factors, neural mediators, secretions, luminal food, and absorbed nutrients all modulate gastric mucosal growth (17). EGF, TGF-α< and insulinlike growth factor directly stimulate gastric mucosal growth (17,18). EGF is ideally suited to participate in gastric repair because it is acid stable and stimulates epithelial migration, DNA synthesis, and gastric mucus production. TGF-α shares 35% homology with EGF and mimics its mitogenic effects (19). EGF and TGF-α also modulate parietal cell function and inhibit gastric hydrochloric secretion (20).
Cell progenitors reside in the mucous neck region giving rise to multiple cell types. One type migrates toward the luminal surface and differentiates into foveolar cells. Other cell lineages migrate downward from the mucous neck region slowly differentiating into parietal, chief, mucous, and endocrine cells (Fig. 4.3). Mature parietal cells and chief cells do not divide. Parietal, chief, and endocrine cells turn over more slowly than surface cells, renewing themselves every 1 to 3 years.
Acid and Pepsin Secretion
Three separate pathways stimulate acid secretion: (a) a neural pathway, which delivers transmitters such as acetylcholine
(ACH) released from postganglionic nerves in the stomach wall; (b) an endocrine pathway, which delivers hormones such as gastrin; and (c) a paracrine pathway, which delivers tissue factors such as histamine (Fig. 4.5) (21). Potentiating interactions between two or three gastric secretagogues amplify oxyntic secretory responses. Histamine released from lamina propria mast cells and from enterochromaffinlike (ECL) cells binds to H2 receptors on oxyntic cells, resulting in up-regulation of cholinergic and gastrin receptors, making them more sensitive to subsequent stimulation by their respective secretagogues. ACH binds to muscarinic cholinergic receptors on oxyntic cells, stimulating acid secretion. ACH in the antral mucosa inhibits somatostatin production, a peptide that inhibits gastrin release (22).
(ACH) released from postganglionic nerves in the stomach wall; (b) an endocrine pathway, which delivers hormones such as gastrin; and (c) a paracrine pathway, which delivers tissue factors such as histamine (Fig. 4.5) (21). Potentiating interactions between two or three gastric secretagogues amplify oxyntic secretory responses. Histamine released from lamina propria mast cells and from enterochromaffinlike (ECL) cells binds to H2 receptors on oxyntic cells, resulting in up-regulation of cholinergic and gastrin receptors, making them more sensitive to subsequent stimulation by their respective secretagogues. ACH binds to muscarinic cholinergic receptors on oxyntic cells, stimulating acid secretion. ACH in the antral mucosa inhibits somatostatin production, a peptide that inhibits gastrin release (22).
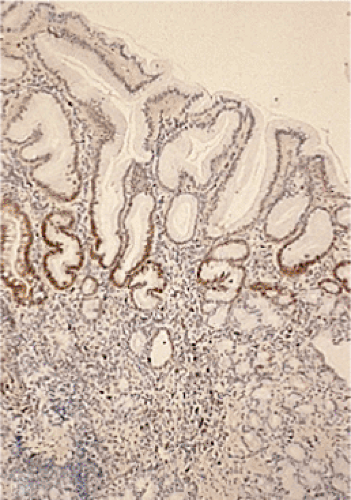 FIG. 4.4. Proliferative zone of the gastric mucosa highlighted by Ki-67 immunostain. |
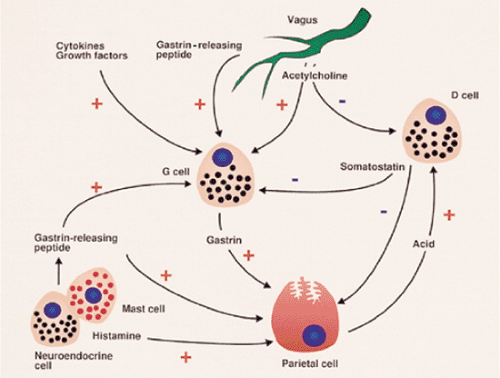 FIG. 4.5. G cells are central to parietal cell secretion. The G cell is positively influenced by acetylcholine release and gastrin-releasing peptide from the vagus as well as from cytokines and growth factors in the gastric mucosa. Mucosal neuroendocrine cells also produce gastrin-releasing peptide, positively influencing the G cell. Vagal stimulation releases acetylcholine, negatively influencing D cells, suppressing somatostatin function. Somatostatin negatively regulates G-cell activity, suppressing gastrin production. G cells, once stimulated, act directly on parietal cells through release of gastrin or indirectly through enterochromaffinlike cells that produce histamine. Histamine released from mast cells or neuroendocrine cells positively influence parietal cells to secrete acid. Somatostatin has a negative influence on acid secretion and forms part of the feedback loop in which acid secretion by parietal cells enhances D-cell function. |
Antral G cells release gastrin when the antrum becomes alkalinized, stimulating acid secretion via gastrin receptors on parietal cells and histamine release from ECL cells (23). Vagal stimuli also liberate gastrin-releasing peptide, prompting G cells to produce gastrin and stimulating ECL cells to release histamine. Pepsinogens synthesized by gastric chief cells (24) have no digestive capacity until they are broken down into pepsin, a reaction that maximally occurs in an acid environment. The stomach produces two immunologically distinct pepsinogens: Pepsinogen 1 (PG1) and pepsinogen 2 (PG2). PG1 is only present in fundic chief and mucous neck cells, whereas PG2 is produced by chief cells, fundic mucous neck cells, cardiac and pyloric glands, and Brunner glands (25). Serum levels of PG1 and PG2 reflect the volume of the cells that produce them. PG1 levels below 20 mg/dL indicate a profound loss of fundic gland volume, as occurs in autoimmune gastritis.
Gastric Motor Functions
In addition to its secretory, digestive, hormonal, and mucosal barrier functions, the stomach has three specific motor functions: (a) storage and volume adaptation, (b) mixing of gastric contents, and (c) forward propulsion of its contents, or gastric emptying. When empty, the stomach is at its smallest possible size. Filling the stomach with fluid or food increases
the gastric luminal volume without increasing gastric pressure. Gastric motility is regulated by the extrinsic nerves and by the intrinsic myenteric plexus, which contains cholinergic nerves, adrenergic nerves, and nonadrenergic, noncholinergic nerves.
the gastric luminal volume without increasing gastric pressure. Gastric motility is regulated by the extrinsic nerves and by the intrinsic myenteric plexus, which contains cholinergic nerves, adrenergic nerves, and nonadrenergic, noncholinergic nerves.
Anatomy
The stomach lies intraperitoneally, extending from the lower end of the esophagus at the Z line at the level of the 11th thoracic vertebrae, crossing to the right of the midline, and ending in the duodenum. The opening that connects the esophagus to the stomach is known as the cardiac orifice; the opening from the stomach to the intestine is known as the pyloric orifice. The stomach has two curvatures: The greater curvature, the inferior border of the stomach, is convex in shape, extending from the gastroesophageal junction to the duodenum. It is more freely movable than the lesser curvature. The concave lesser curvature is the upper margin of the stomach. The size and shape of the usually J-shaped stomach (Fig. 4.6) depends on body position and the degree of filling. Its anterior surface abuts the abdominal wall and the inferior surface of the left lobe of the liver. Posteriorly, it abuts the pancreas, splenic vessels, left kidney, and adrenal gland. The lesser curvature is suspended from the inferior aspect of the liver by the hepatogastric ligament and the lesser omentum. The greater omentum extends caudally from the greater curvature. The gastric fundus touches the dome of the left diaphragm and the upper left margin of the greater omentum rests against the spleen to which it is attached by the gastrosplenic ligament.
The stomach has four layers: Mucosa, submucosa, muscularis propria, and serosa (Fig. 4.7). The gastric wall is slightly firm but pliable and, with the exception of the pylorus, usually does not measure more than 0.5 cm in thickness. The stomach is often divided into four anatomic regions: Cardia, fundus, body, and antropyloric region (Fig. 4.8). The cardia, a narrow, ill-defined region, is not grossly distinctive and is identified histologically by the presence of cardiac glands. Its anatomy is discussed in Chapter 2. The fundus is that portion of the gastric body that protrudes over a horizontal line drawn from the esophagogastric junction (Fig. 4.8). It blends into the gastric body, which constitutes most of the stomach. The body is demarcated from the distal portion, called the pyloric antrum, by a notch in the lesser curvature, the incisura angularis. Numerous longitudinal, grayish pink mucosal folds (called rugae) lie parallel to the lesser curvature (Fig. 4.9) and characterize the mucosa of the gastric body.
 FIG. 4.6. Unopened stomach demonstrating its classic J shape. Esophagus and duodenum is also present. |
 FIG. 4.7. Full-thickness section of the normal stomach. The four layers are easily discerned. |
The triangularly shaped antrum occupies the distal third of the stomach proximal to the pyloric sphincter extending further along the lesser curvature (5 to 8 cm) than along the greater curvature (6 cm), often almost reaching the cardia (26). The antrum is more firmly anchored to the underlying submucosa than the remainder of the stomach. A greatly thickened distal muscular wall forms the pyloric sphincter. A narrow lumen passes through the pyloric sphincter. The pyloric canal, or pyloric channel, measures 2.5 cm in length. The various gastric zones are not fixed anatomic entities; their extent varies between individuals, with age, and with disease processes.
The gastric muscularis propria differs from that of the rest of the gastrointestinal (GI) tract in that it consists of
muscle fibers oriented in three different directions: The outer longitudinal, the middle circular, and the inner oblique layer. Only the middle circular layer is complete. It is the strongest of the three muscle layers and it becomes hypertrophic proximally and distally at the sphincters. The pyloric musculature consists of two layers: A thick inner circular layer and a thin outer longitudinal layer. The muscularis mucosae consists of two or three muscle layers.
muscle fibers oriented in three different directions: The outer longitudinal, the middle circular, and the inner oblique layer. Only the middle circular layer is complete. It is the strongest of the three muscle layers and it becomes hypertrophic proximally and distally at the sphincters. The pyloric musculature consists of two layers: A thick inner circular layer and a thin outer longitudinal layer. The muscularis mucosae consists of two or three muscle layers.
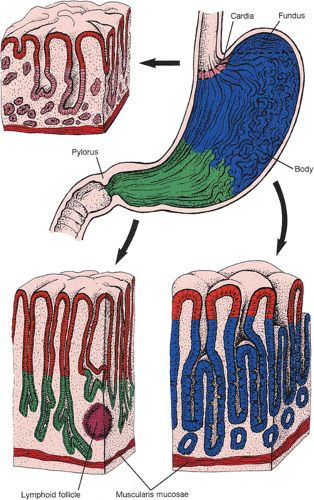 FIG. 4.8. Diagram of the four anatomic and three histologic regions of the stomach. The depths of the gastric pits (red) and the glandular composition are different in the various areas of the stomach. The color of the glands corresponds to the color of the anatomic regions. The histology of the glands differs in the pink, green, and blue areas. The gastric pits are similar (red) throughout the entire stomach. |
 FIG. 4.9. Gastric rugae. When the normal stomach is opened the rugae appear as coarse folds of the mucosa. |
The stomach has a rich blood supply that derives from the celiac, hepatic, and splenic arteries. Mucosal capillaries lie beneath the epithelium. The capillary networks drain into subepithelial venules, which converge into submucosal veins. Venous drainage is through the portal system to the liver. Right and left gastric veins drain the lesser curvature. The left gastric vein arises on the anterior and
posterior gastric surfaces. Esophageal veins enter before it reaches the portal vein. Venous drainage from the anterior and posterior surfaces of the antropyloric region forms the right gastric vein, which empties directly into the portal vein. The abundant blood supply explains why gastric ischemia is unusual and why gastric hemorrhages are so life threatening.
posterior gastric surfaces. Esophageal veins enter before it reaches the portal vein. Venous drainage from the anterior and posterior surfaces of the antropyloric region forms the right gastric vein, which empties directly into the portal vein. The abundant blood supply explains why gastric ischemia is unusual and why gastric hemorrhages are so life threatening.
Gastric lymphatic distribution resembles that of the colon. Lymphatics are absent from the superficial mucosa but are present in the deep interglandular region (27). They converge into thicker channels piercing the muscularis mucosae and enter the larger submucosal plexus. From there, they drain into the lymphatic plexus between the inner and outer layers of the muscularis propria (27). The lymphatic distribution generally follows that of the main arteries and veins. Gastric lymphatics drain into numerous lymph nodes situated in chains along the greater and lesser curvatures, the cardia, and the splenic hilum. There are four drainage areas. The largest drainage area comes from the lower esophagus and most of the lesser curvature (Fig. 4.10). It follows the left gastric artery and drains into the left gastric lymph nodes. The pylorus drains to the right gastric and hepatic lymph nodes (Fig. 4.10). Lymphatics from the cardia drain into pericardial lymph nodes surrounding the gastroesophageal junction, and efferent channels from the left gastric lymph nodes drain into the celiac lymph nodes. The proximal greater curvature drains into the pancreaticosplenic lymph nodes in the splenic hilum. The distal greater curvature drains into right gastroepiploic lymph nodes in the greater omentum and to the pyloric lymph nodes at the pancreatic head. The pyloric portion of the lesser curvature drains into the right gastric lymph nodes, which then drain into hepatic nodes located along the course of the common hepatic artery. Efferents from all four lymph node groups ultimately pass to the celiac nodes around the main celiac axis.
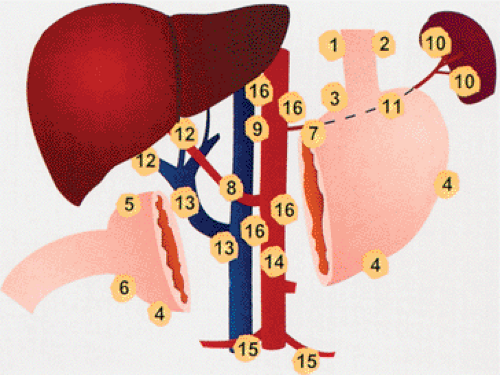 FIG. 4.10. Regional gastric lymph node drainage. The node groups are perigastric nodes (1–6), left gastric (7), along the splenic artery (10,11), along the hepatoduodenal ligament (12), para-aortic (9,16), and intra-abdominal nodes (8,13–15). |
The stomach is innervated by sympathetic and parasympathetic components of the autonomic nervous system as well as by the peptidergic neural system. The parasympathetic nerve supply derives from the vagus and its branches. Numerous neuropeptides produced and released from nerve fibers in the stomach wall regulate gastric function (28).
A thin translucent serosa (the visceral peritoneum) invests the outer portion of the stomach. The serosa normally appears pink-tan, smooth, and glistening.
Normal Gastric Histology
Histologically, the stomach contains three major epithelial compartments: The gastric pits and surface lining, the mucous neck region, and the glands. The nature and relative thickness of the glands and pits (Fig. 4.8) defines each gastric zone. Foveolar (or surface) epithelium lines the entire gastric surface and short, straight, narrow gastric pits (foveolae) that lie parallel to one another. Gastric glands empty into the bottom of the pits. The stomach is divided into the cardiac, oxyntic, and pyloric areas based on its glandular components. The oxyntic mucosa, which secretes acid and pepsinogen, occupies the proximal 80% of the stomach, including the mucosa of the body and fundus. It is thicker than the cardiac and pyloric mucosa due to the presence of specialized acid-secreting glands. Fundic gastric pits are shorter than elsewhere, occupying only 25% of the mucosal thickness (Fig. 4.8). The antropyloric mucosa constitutes the distal 20% of the stomach and contains mucus-secreting glands and endocrine cells. Cardiac mucosa extends distally from the lower esophagus. Transitional zones between the different areas are gradual and junctional mucosa showing mixed histologic features commonly measures up to 1 cm in width. Each of the gastric epithelial cell types produces a specific cell product (Table 4.1).
TABLE 4.1 Gastric Epithelial Cells and Their Products | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||
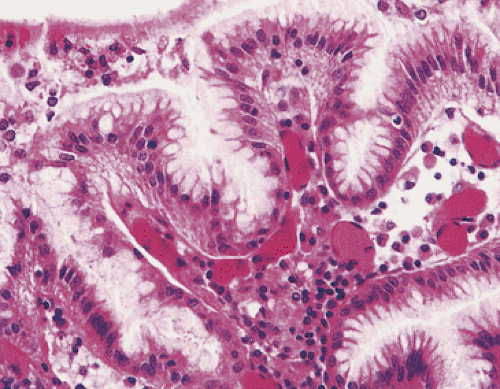 FIG. 4.11. Mucus-secreting foveolar cells cover the gastric surface and line the upper gastric pits. |
Surface Epithelium (Foveolar Epithelium)
Tall, columnar, foveolar epithelium covers the entire gastric mucosa. It consists of a single layer of mucus- and bicarbonate-secreting cells with irregular, basally situated nuclei and a single inconspicuous nucleolus (Figs. 4.11 and 4.12). Ovoid, spherical, mucin-containing, membrane-bound granules pack the supranuclear cytoplasm. The mucin stains strongly with the periodic acid–Schiff (PAS) stain (Fig. 4.13); it is negative or only weakly positive with mucicarmine stains. Numerous spot desmosomes and gap junctions maintain intercellular communication between the surface mucous cells, regulate cell differentiation (29), and help maintain mucosal barrier integrity. The surface mucous cells are produced in the mucous neck region, migrate upward, and extrude from the surface.
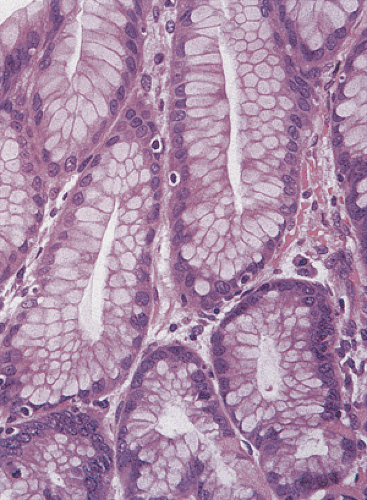 FIG. 4.12. Lining of the gastric pits. Variably mature foveolar cells populate the pits. The mucinous contents increase in size as the cells progress toward the surface. Foveolar epithelia characteristically have basal nuclei with supranuclear mucin collections. |
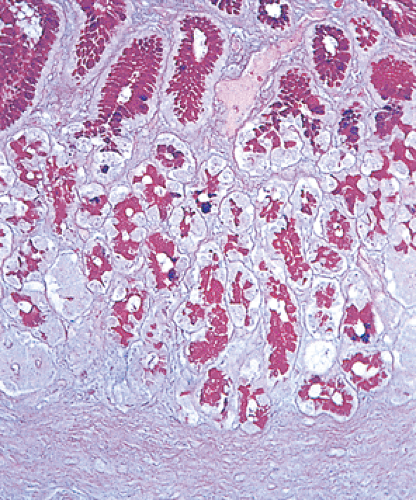 FIG. 4.13. Alcian blue–periodic acid–Schiff stain of cardia. |
Gastric Glands
The three types of gastric glands are fundic, cardiac, and pyloric glands. Cardiac and antral glands contain mucin and are compared in Table 4.2. The cardiac mucosa is discussed in detail in Chapter 2. Both cardiac and pyloric glandular cells have ill-defined borders and a bubbly vesicular cytoplasm containing neutral mucin (Figs. 4.14 and 4.15). Unlike foveolar cells, the mucin fills the basal cytoplasm displacing and flattening the nuclei. Pyloric glands contain two major cell types: Tall columnar cells, which secrete neutral mucin, and scattered endocrine cells. Rare parietal and chief cells may also be present. The oxyntic mucosa characteristically contains long, tightly packed oxyntic glands and short foveolae. In contrast to cardiac and pyloric glands, oxyntic glands are straight rather than coiled. Up to three gastric glands empty into the base of a gastric pit. Oxyntic mucosa contains six different cell types: Surface foveolar cells, isthmus mucous cells, parietal cells, mucous neck cells, chief cells, and endocrine cells (Figs. 4.16 and 4.17). The gland neck contains undifferentiated, mucous neck, and parietal
cells; the glandular bases contain parietal, chief, and endocrine cells.
cells; the glandular bases contain parietal, chief, and endocrine cells.
TABLE 4.2 Comparison of Antral and Cardiac Glands | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Mucous Neck Cells
Mucous neck cells reside in the neck and isthmic region of the gastric glands (Fig. 4.18). They are continuous with, and resemble, foveolar epithelium, but they contain fewer cytoplasmic mucous granules. They derive from mitotically active stem cells in the neck region. These tall, irregularly shaped cells with basal nuclei produce acid glycoproteins, which differ from the neutral mucins secreted by foveolar epithelium. Mucous neck cells may be difficult to recognize in routine sections but they can be highlighted using a PAS stain. The major function of mucous neck cells is mucosal proliferation and regeneration. However, mitoses are rare unless regeneration is occurring.
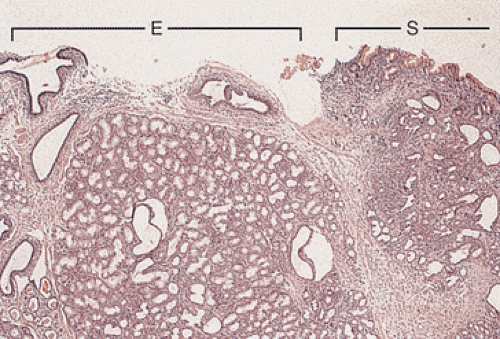 FIG. 4.14. Low magnification showing the junction of the esophagus bracketed by the band labeled E and cardiac glands bracketed by the band labeled S. Submucosal glands lie both underneath the esophageal-lining epithelium and the gastric-lining epithelium. The esophageal lining has been denuded, although major ducts remain. Note that the glandular configuration underlying the S resembles those underlying the E. |
Parietal Cells
Parietal cells constitute approximately one third of the cells in oxyntic glands. They arise from progenitor cells in the lower isthmus and slowly migrate down into the deeper parts of the gland. Intermediate forms exist between immature and mature parietal cells. Parietal cells are easily identifiable by their large size, pyramidal shape, central nuclei, and intensely eosinophilic or clear cytoplasm. Their tapered apical ends tend to bulge into the glandular lumen, whereas their broader basal surfaces rest against the basement membrane (Fig. 4.19). Parietal cells produce hydrochloric acid, intrinsic factor, TGF-α, and cathepsins B and H. In the nonsecretory state, an extensive closed system of smooth membranes, the tubulovesicular system, occupies the cytoplasm adjacent to intracellular canaliculi. Stimulation of acid secretion causes the tubulovesicles to fuse with the canaliculi and the apical secretory membrane, resulting in up to a 40-fold expansion of the apical membrane area. The microvilli become more prominent (30). The canaliculi derive from the smooth endoplasmic reticulum and contain the hydrogen ion pump, a unique H+,K+-ATPase that exchanges H+ for K+ across the apical membrane (31). When acid secretion is inhibited, the situation reverses. The canaliculi collapse, the microvilli recede, and cytoplasmic tubulovesicular structures become prominent again as the cell returns to its resting state.
The basolateral membranes of parietal cells carry receptors for histamine, gastrin, and acetylcholine (Fig. 4.20). Ligand binding to these receptors stimulates hydrogen ion secretion into the lumen and bicarbonate ions into the interstitium. There is a simultaneous appearance of pathways for K+ and Cl– movement coordinated with exchange of H+ for K+ powered by the proton pump located in the membranes. Basolateral uptake of chloride by parietal cells is mediated by an HCO3– Cl– anion exchange mechanism (31).
Chief Cells
Chief (zymogenic) cells arise from isthmic stem cells. These triangular low columnar cells contain a coarsely granular, pale gray-blue, basophilic cytoplasm with one or more small nucleoli. Chief cells constitute 20% to 26% of oxyntic glands lying
deep within them (Fig. 4.19). The basal cytoplasm contains an extensive rough endoplasmic reticulum, which appears as a striated basophilic region. Chief cells produce lipase and pepsinogen, the pepsin precursor. Pepsinogen secretion is stimulated by the same agents that stimulate acid secretion. Secretory granules form in the Golgi complex and are released by exocytosis. Zymogenic cells degenerate via necrosis or apoptosis. Apoptotic cells are phagocytosed by neighboring zymogenic cells or by lamina propria macrophages that break through the basement membrane of oxyntic glands.
deep within them (Fig. 4.19). The basal cytoplasm contains an extensive rough endoplasmic reticulum, which appears as a striated basophilic region. Chief cells produce lipase and pepsinogen, the pepsin precursor. Pepsinogen secretion is stimulated by the same agents that stimulate acid secretion. Secretory granules form in the Golgi complex and are released by exocytosis. Zymogenic cells degenerate via necrosis or apoptosis. Apoptotic cells are phagocytosed by neighboring zymogenic cells or by lamina propria macrophages that break through the basement membrane of oxyntic glands.
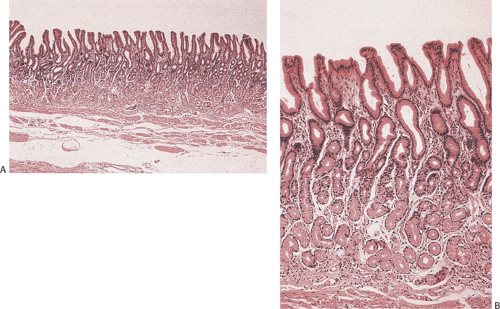 FIG. 4.15. Normal antral glands. A: Low magnification of a normal antrum. B: Higher magnification showing the mucous glands. |
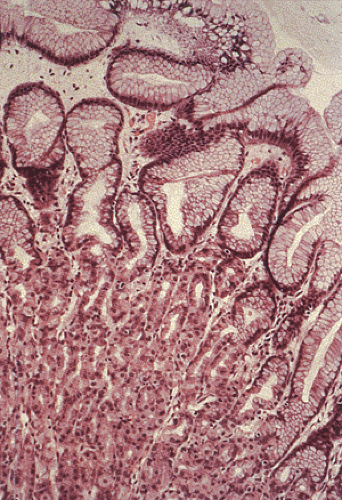 FIG. 4.16. Normal oxyntic mucosa. The bottom portion of the photograph contains densely packed oxyntic glands containing chief cells, parietal cells, and endocrine cells. |
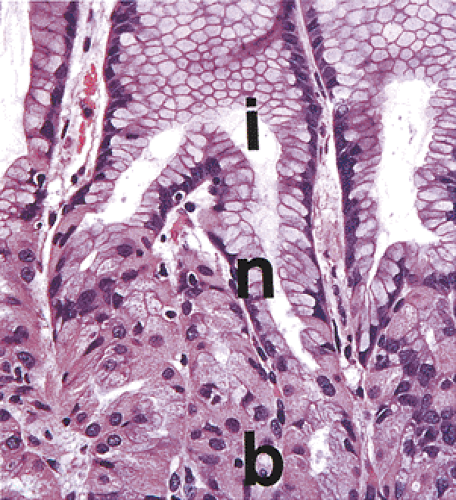 FIG. 4.17. Oxyntic gland showing its base (β), neck (n), and isthmus (i). |
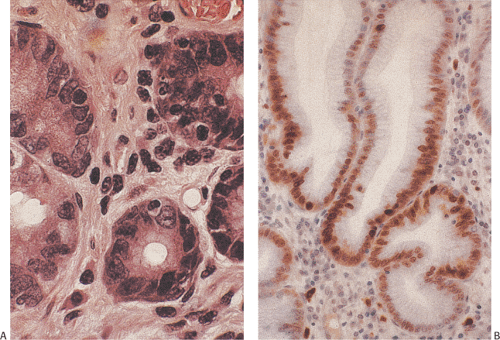 FIG. 4.18. Mucous neck cells. A: High magnification of the mucous neck region showing the presence of tall cells lacking the differentiated features of either foveolar epithelium or underlying glandular epithelium. Mitoses are absent. B: Ki-67 immunostaining showing the proliferative nature of this region. |
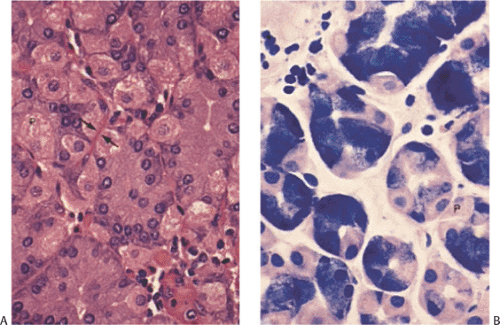 FIG. 4.19. Normal oxyntic glands. A: The plump eosinophilic cells are parietal cells, and the chief cells are basophilic. Note the prominent capillaries lying beneath the parietal cells (arrow). B: Oxyntic mucosa stained with Giemsa. The parietal cells (P) are particularly prominent. |
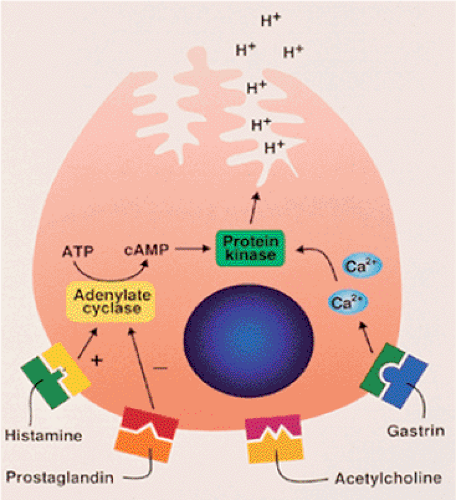 FIG. 4.20. Parietal cells have various receptors along their basolateral membranes. These include those for histamine, prostaglandin, acetylcholine, and gastrin. Ligands bind their respective receptors and activate protein kinase through cyclic adenosine monophosphate (cAMP). The process involves calcium ions. These events result in cellular secretion of H+ ions. ATP, adenosine triphosphate. |
Antral and Pyloric Glands
In the antral and pyloric region, the pits occupy approximately 40% of the mucosa. These branch and may not always lie perpendicular to the surface. The deep mucosa contains coiled tubular glands that are lined by faintly granular mucin-secreting cells, often resembling mucous neck cells.
Endocrine Cells
The stomach contains a prominent and diverse endocrine cell population that is discussed further in Chapter 17.
Gastric Transitional Zones
Gastric transitional zones are the junctional zones between the different types of mucosae: Antral/body, body/cardia, and antrum/duodenum. They are dynamic areas that involve a gradual merging of mucosal types so that it may be difficult to determine the exact location of each of the transitional zones. The antral/body transitional zone is usually located approximately two fifths of the way along the lesser curvature; on the greater curvature it is closer to the pylorus (26). The most useful criteria to determine when one crosses from body into antrum are the absence of chief cells and the change from single tubular glands in the body to branched glands in the antrum (32). It may be difficult to determine the site of origin of the gastric mucosa, particularly when it is altered by inflammation, atrophy, and/or metaplasia. In particular, it may be difficult to differentiate between a nonatrophic antral gastritis and an atrophic body gastritis with pyloric metaplasia.
Lamina Propria and Mononuclear Cells
The surface, pits, and glands are supported by a well-developed lamina propria that contains a fine reticulin meshwork with occasional collagen and elastic fibers condensed beneath epithelial basement membranes and blood vessels. The lamina propria is more abundant in the superficial mucosa between the pits than in the lower mucosa. It contains numerous cell types including fibroblasts, macrophages, plasma cells, and lymphocytes. The lymphocytes are predominantly immunoglobulin (Ig) A–producing B cells. IgG- and IgM-secreting cells are also present. Intraepithelial T lymphocytes are present but they are much less frequent than in the small bowel. These may have a perinuclear halo superficially resembling endocrine cells. There are also a small number of lamina propria T cells, neutrophils, and mast cells. Lymphoid follicles suggest the diagnosis of chronic gastritis. The lamina propria also contains capillaries, arterioles, and nonmyelinated nerve fibers.
Lymphatics appear in the deep lamina propria adjacent to and within the muscularis mucosae. The upper and mid–lamina propria lacks lymphatics. In contrast, the entire mucosa contains a rich supply of capillaries, many of which lie adjacent to the basal lamina of the gastric glands and surface epithelium.
Neuromuscular Relationships
The muscularis mucosae varies from 30 to 200 μm in thickness. It becomes hyperplastic and extends into the overlying mucosa in certain conditions as discussed later. The muscularis propria consists of smooth muscle cells and contains nerve fibers and a myenteric plexus. There are also interstitial cells of Cajal (ICC), which have contacts with each other and with the smooth muscle cells and nerve endings in the muscularis propria (33). They lie in the myenteric plexus and in the circular muscle (33). They serve as gastric pacemakers.
Serosa
The serosa, the outermost gastric layer, consists of connective tissue and a mesothelial lining continuous with the peritoneum.
Structural Abnormalities
Duplications
Gastric duplications constitute only 3.8% to 10% of all gastrointestinal duplications (34). They affect females more commonly than males. Sixty-five percent of patients present during the first year of life, often with respiratory distress or as an intrathoracic or extragastric mass (34). Occasionally, the lesions present in adults (35). Thirty-five percent of patients have associated developmental anomalies (Table 4.3) (36). Complications include ulceration, bleeding, rupture, fistula formation, and, rarely, carcinoma (37). Distal duplications cause gastric outlet obstruction, pain, vomiting, fever, weight loss, or hemorrhage (34).
Gastric duplications appear as intramural cylindrical or cystic masses ranging in size from 1.3 to 12 cm. They share a common blood supply with the rest of the stomach; a common musculature invests them. Most duplications occur on the greater curvature (34); one third affect the distal stomach. They may be complete or incomplete, communicating or noncommunicating. Alimentary mucosa lines gastric duplications. This lining resembles and/or differs from that of the normal stomach. Gastric and small intestinal epithelium may coexist within a single duplication. Pancreatic tissue may also be present, as may respiratory mucosa, cartilage, or submucosal glands.
Dextrogastria
In patients with situs inversus, the stomach lies to the right of the midline, a condition known as dextrogastria. The esophageal diaphragmatic hiatus also lies on the right side; the first part of the duodenum lies on the left side. Dextrogastria affects approximately 1 of every 6,000 to 8,000 births (38). Situs inversus affecting only the stomach and duodenum (with the remainder of the thoracic and abdominal viscera lying in their normal positions) is extremely rare (38). The stomach either lies completely behind the liver or above it. Although abnormally positioned, gastric structure and function are normal.
Gastroschisis
The incidence of gastroschisis increased from 0.006 per 1,000 in 1968 to 0.089 per 1,000 in 1977. Young, socially disadvantaged women have the highest risk of giving birth to a child with gastroschisis (39). Gastroschisis presumably results from vascular injury to the abdominal wall causing defective somatopleural mesenchymal differentiation during the 5th to 11th fetal weeks (40). Gastroschisis may complicate premature atrophy or abnormal persistence of the right umbilical vein (40). Portions of the stomach, small intestine, and colon herniate through an abdominal wall defect lateral to the umbilicus. Since no peritoneal sac or sac remnant covers the eviscerated abdominal contents, the herniated organs are exposed to amniotic fluid leading to gastric wall thickening, serosal edema, and fibrinous exudates.
TABLE 4.3 Lesions Coexisting with Gastric Duplications | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Congenital Hiatal Hernia
A congenital periesophageal gap or congenital elongated esophageal hiatus can result in a congenital hiatal hernia with invagination of abdominal contents into the thorax (Fig. 4.21). The defect results from failure of the pleuroperitoneal folds to develop or from failure of the pleuroperitoneal canal to close. Urinary tract abnormalities are common in patients with congenital posterolateral diaphragmatic defects including renal agenesis, dysplasia, hypoplasia, or hydronephrosis.
Acquired Hiatal Hernia
The freely movable stomach can prolapse through natural and surgically created diaphragmatic defects, coming to lie in the thoracic cavity. Clues that a biopsy may come from the area of a hiatal hernia include the presence of variably inflamed cardiac or oxyntic mucosa with edema, lymphatic dilation, and pronounced muscle hyperplasia, splaying, or stranding. Squamous metaplasia may be present.
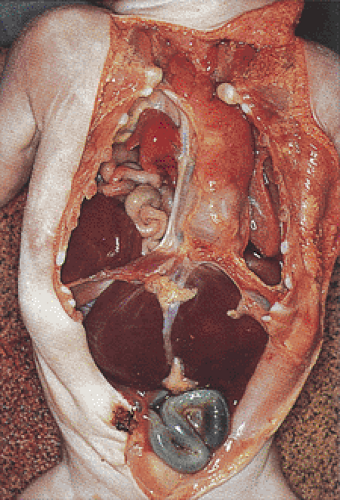 FIG. 4.21. Congenital right diaphragmatic defect. The small intestine has herniated into the right thoracic cavity with partial collapse of the right lung and deviation of the trachea to the left. A large thymus overlies the trachea. |
Diverticula
Congenital Diverticula
Gastric diverticula are rare, ranging in incidence from 0.02% to 0.18% (41). Most arise on the posterior wall, in a juxtacardiac position (41). Congenital diverticula appear as solitary, sharply defined, round, oval, or pear-shaped pouches communicating with the gastric lumen via a narrow or broad-based mouth (Fig. 4.22).
Acquired Diverticula
Acquired diverticula almost always originate in the distal stomach as a complication of antral inflammation. Fibrosis following acute inflammation causes traction on the tissues and mucosa herniates through the gastric wall. Therefore, antral diverticula should be carefully evaluated to exclude the presence of an underlying pathologic process such as gastritis, peptic ulcer disease, or neoplasia.
Atresia, Webs, and Diaphragms
Congenital gastric outlet obstruction due to a membranous antral web is extremely rare with an incidence of 0.0001% to 0.0003% of live births (42). A high incidence of associated extraintestinal anomalies and a strong familial history for pyloric atresia support the theory that atresias and webs result from underlying genetic alterations. Polyhydramnios affects more than 50% of cases. Gastric atresia may associate with trisomy 21, epidermolysis bullosa, or esophageal and anal atresia (43,44). Hereditary multiple gastrointestinal atresias affect the GI tract from the pylorus to the rectum and are inherited in an autosomal recessive fashion. They may associate with immunodeficiency syndromes (44,45).
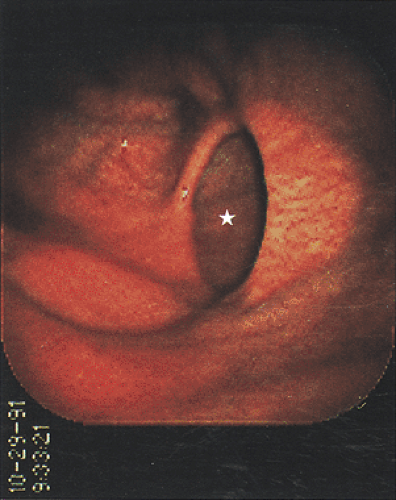 FIG. 4.22. Gastric diverticulum. Endoscopic appearance. The orifice of the diverticulum is indicated by a star. Note the similarity of the mucosal lining in both the native stomach as well as in the diverticulum. |
Most patients with gastric atresia present in the first few days of life with bile-free vomiting and abdominal distention. Type 1 gastric atresia (the most common type) consists of an internal web or diaphragm that completely separates the stomach from the duodenum. Type 2 atresia (the rarest type) consists of a thin, fibrous cord that connects a blind gastric pouch to a distal blind small intestinal segment. Type 3 atresia consists of a blind gastric pouch and a blind distal intestinal pouch without intervening tissue (44,45).
A variably inflamed distal antral mucosal fold with a central aperture measuring from 1 to 10 mm in diameter lies perpendicular to the long axis of the antrum. The serosa may appear indented at the level of the diaphragm. Antral mucosa lines both sides of the diaphragm covering a submucosal core. Heterotopic pancreatic tissue sometimes lies within webs and diaphragms (44,45). In adults, diaphragms and webs complicate inflammatory conditions (35).
Pyloric Stenosis
Pyloric stenosis affects both children and adults and assumes several forms.
Infantile Hypertrophic Pyloric Stenosis (Congenital Pyloric Stenosis)
This disorder is discussed in Chapter 10 since it is primarily a motility-related disorder.
Acquired Pyloric Stenosis
Adult forms of hypertrophic pyloric stenosis result from inflammation associated with antral gastritis and/or peptic ulcer disease, or from inherent neuromuscular abnormalities. Partial gastric obstruction leads to an increased stomach size and weight due to localized or diffuse gastric muscular hypertrophy and hyperplasia, increased mucosal thickness, and G-cell hyperplasia. The pyloric deformity leads to bile reflux and secondary alkaline reflux gastritis.
Torus Hyperplasia
The very rare condition known as focal pyloric hypertrophy (torus hyperplasia) appears as a localized area of circular muscle hypertrophy affecting the lesser curvature near the pyloric torus. The lesion may represent a form of acquired pyloric stenosis or it may represent persistence of congenital pyloric stenosis into adulthood. Some speculate
that the lesion results from chronic gastritis or from repeated spastic pyloric contractions (46).
that the lesion results from chronic gastritis or from repeated spastic pyloric contractions (46).
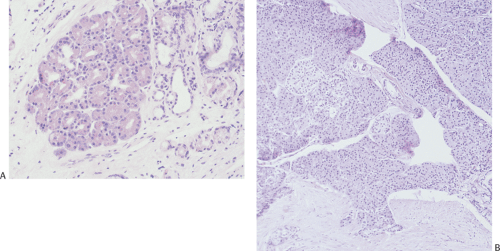 FIG. 4.23. Pancreatic metaplasia versus heterotopic pancreas. A: Pancreatic metaplasia consists of pancreatic acini that merge with the surrounding gastric mucosa. B: Heterotopic pancreas usually involves the submucosa and may contain pancreatic lobules, islets, and ductules (not shown). |
Heterotopias
Normal tissues lie in abnormal locations due to a congenital heterotopia or secondary to a metaplasia. Congenital heterotopias differ from metaplastic (acquired) lesions in that they usually retain a normal organizational structure, whereas metaplastic processes tend to consist of a single cell type lacking normal tissue patterns (Fig. 4.23). Congenital heterotopias result from cellular entrapment during embryonic morphogenetic movements. The congenitally displaced tissues then differentiate along the lines of normal organs in response to the local environment.
Heterotopic Pancreas
Heterotopic pancreas is the most common gastric heterotopia. It accounts for 25% to 30% of all pancreatic heterotopias (47). It is usually an incidental finding, commonly found in the antrum, and followed by the pylorus, greater curvature, and esophagogastric junction. If a tumor or pancreatitis develops, the heterotopic tissue may become symptomatic. Heterotopic pancreas usually appears as a solitary submucosal, hemispheric, umbilicated mass measuring 0.4 to 4.0 cm in diameter. The entry of single or multiple ducts into the gastric lumen produces a symmetric cone or short, cylindric, nipplelike projection (Fig. 4.24). Heterotopic pancreas may also present as large submucosal mucinous cysts. Multiple or pedunculated pancreatic heterotopias are uncommon. Approximately 75% of pancreatic heterotopias lie in the submucosa (Fig. 4.25), with the remainder involving the muscularis propria. Cross section of larger lesions reveals the typical tan, lobulated tissue characteristic of eutopic pancreas. The deeper the lesion, the more disorderly it tends to appear. The pancreatic lobules contain variable mixtures of pancreatic acini, ducts, islets, glands resembling Brunner glands, and hypertrophic smooth muscle fibers. The islets contain variable numbers of pancreatic polypeptide and insulin-producing cells (48). If only pancreatic acini are present, the lesion may represent foci of pancreatic metaplasia (see below), especially if the cells lie within the mucosa. Sometimes one sees both heterotopic pancreatic and gastric tissue lying side by side in the submucosa (Fig. 4.26).
Heterotopic pancreatic tissue does not pose a diagnostic problem when both pancreatic acini and ducts are present. However, lesions containing only smooth muscle and/or pancreatic ducts have been misinterpreted as adenomyomas. A clue that the lesion represents heterotopic pancreatic tissue, rather than an adenomyoma, is the finding of hypertrophic circular and longitudinal smooth muscle cells arranged circumferentially around the ducts in a more or less normal fashion (Fig. 4.25). Secondary changes such as pancreatitis, cyst formation, or neoplasia (islet cell tumors, ductal dysplasia [Fig. 4.27], and adenocarcinoma) may also cause confusion, particularly when they distort the underlying tissue (49). Dilated ducts forming submucosal mucin pools containing epithelial clusters and variable degrees of inflammation, without obvious pancreatic tissue adjacent to the mucin, may suggest a diagnosis of colloid carcinoma. One should not
make a diagnosis of invasive cancer in the absence of significant cytologic atypia and stromal desmoplasia.
make a diagnosis of invasive cancer in the absence of significant cytologic atypia and stromal desmoplasia.
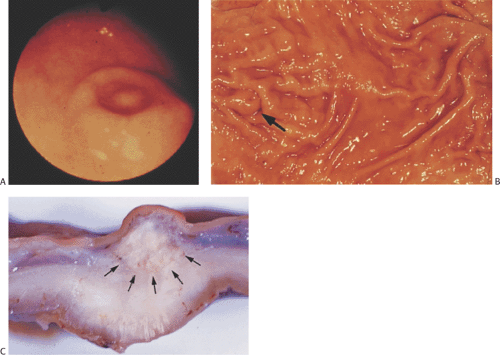 FIG. 4.24. Heterotopic pancreas. The heterotopic pancreas produces a well-defined submucosal mass that is visible endoscopically (A) as well as grossly (B). The submucosal mass distorts the gastric folds (arrow) and appears as a hemispheric lesion with a central umbilication. C: Cross section of the gastric wall demonstrating the presence of a whitish, firm mass lying within the submucosa as indicated by the arrows. |
Heterotopic Gastric Glands
Diffuse or localized submucosal gastric heterotopias occur in up to 14% of stomachs (50). These are either congenital in origin or represent areas of gastritis cystica profunda (discussed below). Congenital gastric heterotopia usually contains oxyntic mucosa with foveolar epithelium arranged in a normal architectural pattern.
Heterotopic Brunner Glands
Heterotopic Brunner glands can accompany heterotopic pancreas, or the heterotopia may contain only Brunner glands and smooth muscle. The heterotopic glands lie in the pylorus and gastric antrum and histologically resemble duodenal Brunner gland hyperplasia (see Chapter 6).
Double Pylorus
Double pylorus is an acquired condition in patients with peptic ulcer disease. Prepyloric ulcers penetrate the pyloric wall, perforate into the duodenum, and create a new mucus-lined channel. Rare examples of congenital double pylorus also exist (51).
Pyloric Mucosal Prolapse
Antral mucosa may prolapse into the duodenum, sometimes forming a mushroom-shaped duodenal or gastric pseudopolyp (Fig. 4.28). It occurs sporadically or complicates gastritis or previous gastric surgery. Submucosal edema (as the result of an underlying inflammation) predisposes to mucosal prolapse. As the edema increases, the tissues fail to return to their normal position; progressive gastric outlet obstruction develops. Patients with mucosal prolapse usually develop crampy abdominal pain, delayed gastric emptying, or vomiting due to the gastric outlet obstruction. The prolapsed mucosa appears variably inflamed, edematous, and necrotic, depending on the duration and severity of the obstruction and the degree of vascular compromise. Pit hyperplasia, pit distortion with cystic serrated branched villiform surfaces, a hyperplastic muscularis mucosae with a muscular lamina propria, erosions, ulcers, glandular atrophy, and variable inflammation may all develop.
Volvulus
Gastric volvulus, also known as gastric torsion, affects both children and adults, usually in the presence of a left
diaphragmatic abnormality (52,53). It presents acutely or chronically. Acute presentations include hemorrhage, ischemia, and infarction (52,53). Most patients are elderly with a chronic form of the disease. They have recurrent epigastric pain, vomiting, and occasional hematemesis.
diaphragmatic abnormality (52,53). It presents acutely or chronically. Acute presentations include hemorrhage, ischemia, and infarction (52,53). Most patients are elderly with a chronic form of the disease. They have recurrent epigastric pain, vomiting, and occasional hematemesis.
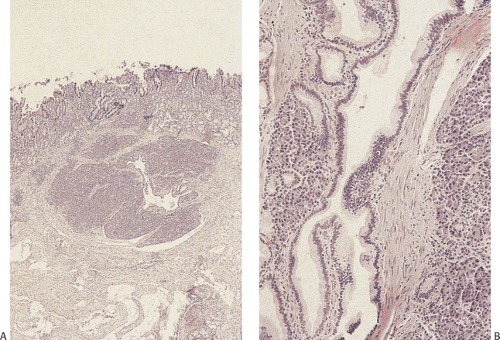 FIG. 4.25. Heterotopic pancreas. A: A lobulated submucosal glandular structure with a central duct. Delicate strands of fibrovascular tissue separate individual lobules. B: Higher magnification of the duct and pancreatic acini on either side of the duct. Prominent smooth muscle fibers also surround the duct. |
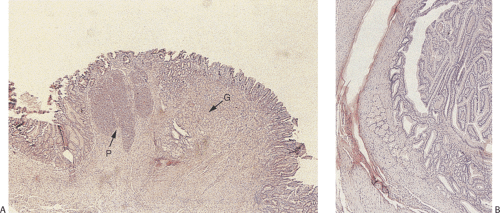 FIG. 4.26. Heterotopic pancreas combined with heterotopic gastric tissue. A: Low-power magnification demonstrating the side-by-side arrangement of heterotopic pancreatic tissue (P) (arrow) and gastric foveolar epithelium (G) (arrow). B: Higher magnification demonstrating the histologic features of the gastric epithelium. |
Gastric volvulus occurs in several forms. The most common type, organoaxial volvulus, accounts for approximately 60% of cases. The stomach twists around the longitudinal axis of its lesser curvature, causing the stomach to turn upside down (Fig. 4.29), producing both proximal and distal
obstructions. Anterior rotation is more common than posterior rotation. Mesenteroaxial volvulus represents 30% of cases. It occurs around a line that runs from the center of the greater curvature to the porta hepatis. Mesenteroaxial and organoaxial volvulus can coexist. The stomach can also twist about the vertical axis of the gastrohepatic omentum producing torsion rather than a true volvulus. As a volvulus develops, the stomach progressively distends, due to accumulated secretions that cannot pass forward or be regurgitated because the volvulus produces both distal and proximal obstruction. Death results from the sequence of obstruction, strangulation, and ischemic necrosis, the latter resulting from compression of the gastric vasculature.
obstructions. Anterior rotation is more common than posterior rotation. Mesenteroaxial volvulus represents 30% of cases. It occurs around a line that runs from the center of the greater curvature to the porta hepatis. Mesenteroaxial and organoaxial volvulus can coexist. The stomach can also twist about the vertical axis of the gastrohepatic omentum producing torsion rather than a true volvulus. As a volvulus develops, the stomach progressively distends, due to accumulated secretions that cannot pass forward or be regurgitated because the volvulus produces both distal and proximal obstruction. Death results from the sequence of obstruction, strangulation, and ischemic necrosis, the latter resulting from compression of the gastric vasculature.
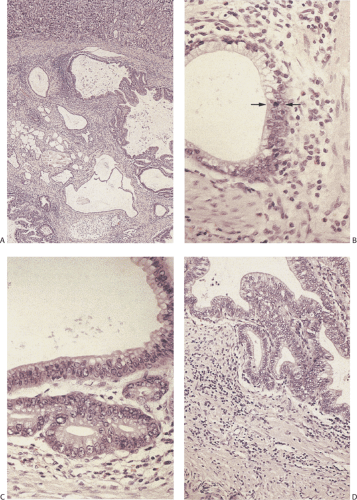 FIG. 4.27. Heterotopic pancreas with dysplastic epithelium. A: Numerous cystic structures lie within the gastric wall. Some are lined by flattened epithelium and contain prominent mucinous collections. Others are lined by benign neoplastic epithelial cells. B: Higher magnification of one of the glands showing the junction of more or less normal epithelium with basal nuclei above the arrows with the benign neoplastic hyperchromatic epithelium with an increased nuclear:cytoplasmic ratio and prominent nucleoli beneath the arrows. C: Higher magnification of the neoplastic epithelium showing cytologic atypia, nuclear stratification, and prominent nucleoli. D: Another area with a more complicated glandular architecture. Note the absence of invasion into the surrounding tissues and the absence of a desmoplastic response. The glands are still surrounded by intact smooth muscle fibers. |
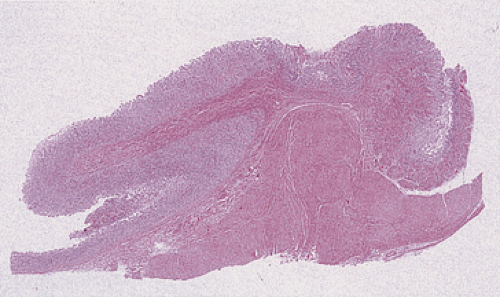 FIG. 4.28. Mucosal prolapse. The prolapsed mucosa assumes a mushroom-shaped configuration overlying the area of the pylorus. |
Microgastria
Microgastria, a rare congenital anomaly, often coexists with other anomalies such as midgut malrotations, cardiac abnormalities, and asplenia (54). The underdeveloped lower esophageal sphincter becomes incompetent and gastroesophageal reflux develops. Symptoms appear in infancy and include failure to thrive, vomiting, and recurrent aspiration pneumonia. Barium swallows demonstrate a small tubular stomach. Histologically, the gastric wall appears hypoplastic (55). The stomach is small, often nonrotated without a clear definition of the various zones. The disorder may result from failed development of the mesogastrium.
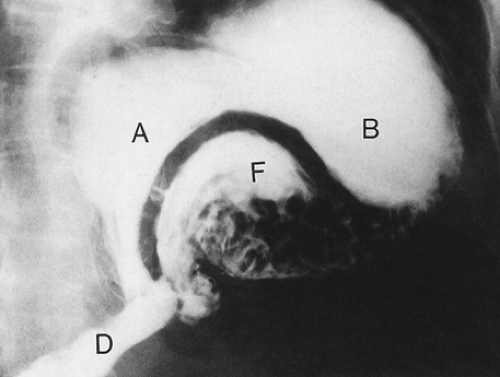 FIG. 4.29. Barium examination showing a gastric organoaxial volvulus. Large thick radiolucent curvilinear defect represents the superiorly located lesser curvature wall and the inferiorly located greater curvature wall. F, fundus; B, body; A, antrum; D, duodenum. |
Mucosal Biopsy
Endoscopic examination with mucosal biopsy and/or cytologic sampling is regularly employed for the initial identification and monitoring of patients with various gastric conditions including gastritis, gastric atrophy, peptic ulcer disease, and neoplastic proliferations. Gastric biopsies are also commonly used to evaluate the stomach for the presence or absence of Helicobacter pylori (HP). Routine gastric biopsies may also show special forms of gastritis (eosinophilic, lymphocytic, and granulomatous), giant fold disease, or polyps. Gastric biopsies can provide information about the grade, extent, and topography of gastritis-related and atrophy-related lesions, information that provides the opportunity to assess the patient’s risk for developing gastric carcinoma. As noted previously, gastric histology varies from area to area and since many gastric diseases are patchy in their distribution, an adequate evaluation often requires examining biopsies from the body, antrum, and any endoscopically visible lesions. The Sydney system requires that biopsies be obtained from five locations in the stomach (the greater and lesser curvature in the antrum, the greater and lesser curvature in the corpus, and the incisura) (56), although this seldom happens in routine clinical practice.
Typically gastroenterologists take a couple biopsies from the antrum and a couple from the corpus. Ideally these should be submitted in separate containers, although this does not always happen either.
Typically gastroenterologists take a couple biopsies from the antrum and a couple from the corpus. Ideally these should be submitted in separate containers, although this does not always happen either.
It is important to note that endoscopic procedures may cause variable degrees of edema, vascular dilation, focal lamina propria hemorrhage, and surface cell flattening. These changes are usually easily distinguishable from mucosal disease because of the absence of both epithelial degeneration and acute inflammation. A systematic examination of gastric biopsies facilitates the diagnosis of various gastric diseases and provides the discipline of establishing a differential diagnosis so that specific diagnostic entities are considered and important entities are not overlooked. One should determine where the biopsy comes from by looking at various mucosal components. However, it may be difficult to determine the precise location of a biopsy because of the presence of atrophy and/or metaplasia. Gastric epithelium may appear in the duodenum in peptic duodenitis; intestinal metaplasia occurs in the setting of gastritis and Barrett esophagus, and pyloric glands develop in the proximal stomach in certain forms of gastritis.
One can broadly divide gastric inflammatory diseases into gastritis and gastropathy. The major distinction between the two is the presence of inflammation in gastritis and its absence in gastropathies. Gastritis typically results from infections, autoimmune or hypersensitivity reactions, or drugs. Determining whether gastritis is a pangastritis or affects only the antral or corpus, whether it is focal or diffuse, and whether it is superficial or occupies the entire mucosal thickness helps distinguish among these etiologies. Other changes that may be assessed include a determination of whether any of the following are present: Surface epithelial damage, superficial stromal hemorrhage, metaplasia (intestinal, pancreatic, or antral), endocrine cell hyperplasia, intraepithelial lymphocytosis, granulomas, apoptosis, or microorganisms. Gastropathies result from hypovolemia, stress, ischemia, drug or alcohol ingestion, chronic congestion, or alkaline reflux from the duodenum. Diagnostic approaches we find useful are shown in Figures 4.30 and 4.31. We do not believe that it is necessary to routinely order special stains to determine whether HP infections or intestinal metaplasia are present, a view shared by others (57). A more detailed evaluation can be used by applying the Sydney system as discussed further in a later section.
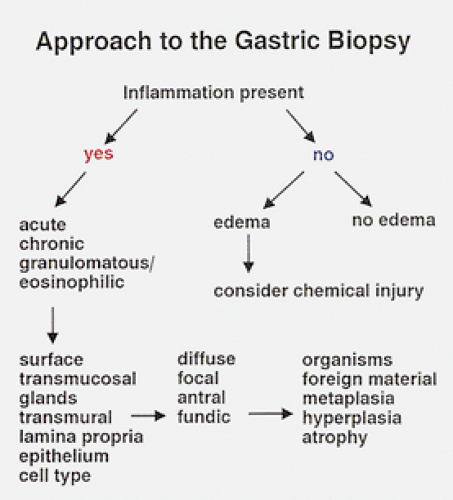 FIG. 4.30. Diagnostic algorithm. One can use this approach once one has determined the subsite localization for the changes that are present. One makes an assessment as to whether or not inflammation is present and, if so, what type and where it is. If inflammation is absent, then one might consider a chemical injury. |
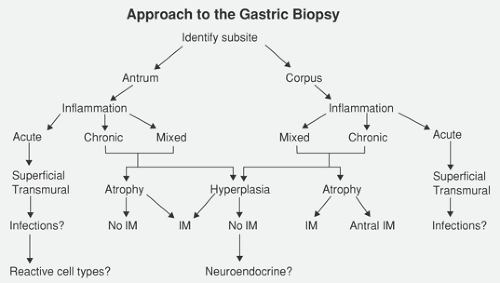 FIG. 4.31. Diagnostic approach to interpreting gastric biopsies. In this approach, one starts by identifying the portion of the stomach that is primarily affected because different entities preferentially affect the antrum and corpus. One then goes through a decision tree based on the character of the inflammation and whether the mucosa appears normal, atrophic, or hyperplastic and contains other cell types. IM, intestinal metaplasia. |
TABLE 4.4 Causes of Acute Gastritis | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||
Acute Gastritis
Acute gastritis (accompanied by acute mucosal injury) results from many disorders with multifactorial etiologies and diverse histologic patterns (Table 4.4). The clinical symptoms, endoscopic findings, and histologic features rarely correlate with one another due to the nonspecificity of the symptoms, the diverse etiologies, and the diffuseness (or focality) of the process. Acute gastritis appears hemorrhagic, nonhemorrhagic, erosive, or nonerosive.
Acute Hemorrhagic, Erosive Gastritis (Stress Gastritis)
Acute erosive gastritis complicates major physiologic disturbances including sepsis, extensive burn injury, head injury, severe trauma, and multiorgan failure. It also develops following ingestion of nonsteroidal anti-inflammatory drugs (NSAIDs), aspirin, or alcohol. Acute gastritis often presents as abdominal discomfort, pain, heartburn, nausea, vomiting, and hematemesis. Bleeding begins 3 to 7 days following a stressful event. It ranges from occult blood loss to massive hemorrhage that originates from innumerable foci of mucosal damage to smaller, more discrete ulcers (Fig. 4.32). Curling ulcers develop in severe burn patients within 24 to 72 hours, predominantly in the proximal stomach.
Pathophysiology
Major factors implicated in the development of stress ulcers include hyperchlorhydria and decreased mucosal protection. The latter results from decreased mucus secretion, mucosal blood flow, and DNA and prostaglandin synthesis, and mucosal barrier breakdown. Indeed, mucosal ischemia is the common denominator of stress-associated injury. Cardiac dysfunction, hemorrhage, shock, and sepsis redistribute blood flow away from the subepithelial capillaries, causing mucosal hypoxia. The hypoxia may persist after recovery from the initial injury, especially as mucosal arterioles contract, further reducing tissue oxygenation (58). An adequate microcirculation that provides nutrients and removes waste products, particularly oxygen-free radicals, is required to maintain the mucosal barrier. A damaged mucosal barrier allows back-diffusion of acid, resulting in tissue acidosis, vascular compromise, mucosal congestion, and necrosis. The mucosal injury increases significantly during the reperfusion that follows ischemia due to the production of toxic oxygen-free radicals (59) by infiltrating neutrophils (60). In addition, activated leukocytes release mediators that reduce mucosal blood flow and increase vascular permeability (61,62). The oxygen-free radical-induced injury is further enhanced by mucosal depletion of the endogenous antioxidant reduced glutathione (GSH) (63). The GSH oxidation/reduction cycle, catalyzed by glutathione peroxidase, reduces H2O2 and breaks the chain reaction that generates highly reactive hydroxyl radicals. GSH acts as a natural scavenger whose superoxide anion protects proteins against oxidation. GSH also plays a major role in restoring other free radical scavengers and antioxidants such as vitamins E and C to their reduced state (64). Prostaglandins limit the initial injury (65). Oxidative stress leads to epithelial growth factor receptor (EGFR) phosphorylation and increased production
of its ligands, EGF and amphiregulin (66). A mucoid cap promotes mucosal restitution by protecting the lamina propria from luminal acid, limiting the extent of the injury (65).
of its ligands, EGF and amphiregulin (66). A mucoid cap promotes mucosal restitution by protecting the lamina propria from luminal acid, limiting the extent of the injury (65).
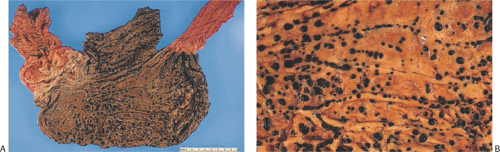 FIG. 4.32. Stress ulcers. A: Multiple small punctate hemorrhagic ulcers in a patient who died of severe head trauma. Scattered small petechiae are also visible. B: Higher power magnification of individual lesions. |
Various factors contribute to the repair of acute gastric mucosal injury. Re-epithelialization requires epithelial migration across an intact basal lamina. This occurs within minutes to hours of the injury to ensure quick restoration of surface epithelial continuity and inhibiting acid back-diffusion (65). In addition, cells in the mucous neck region migrate out of the proliferative zone and progressively differentiate into mature foveolar cells. Gastric mucosal blood flow increases (65). If this is inhibited, mucosal cytoprotective events fail and the mucosal injury progresses with deeper ulceration than would otherwise result from the initial injury.
Pathologic Features
Erosive gastritis and stress ulcers typically appear as multiple lesions located anywhere in the stomach, although they tend to predominate in the oxyntic mucosa (Fig. 4.32). When severe, they extend to the antrum. Stress ulcers tend to be superficial and usually measure <15 mm in diameter. The ulcer bases appear grayish yellow and hemorrhagic with slightly raised, congested, regenerative margins. The intervening gastric mucosa appears diffusely congested and contains numerous small petechial hemorrhages. Alternatively, the mucosa is diffusely hemorrhagic without discrete areas of damage. Early lesions center around intensely congested blood vessels, which leak blood into the surrounding tissues (Fig. 4.33). Extensive hemorrhagic mucosal erosions or ulcers develop in more severe cases. Rare cases present with deep linear ulcers coexisting with more discrete, round, superficial lesions. Curling and Cushing ulcers tend to be deep and single.
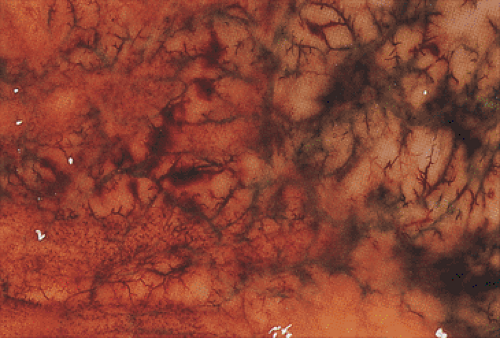 FIG. 4.33. Close-up gross photograph of the gastric mucosa in a patient who died of multiorgan failure. Note the intensely congested vessels as well as the areas of neovascularization. Pinpoint hemorrhages extend from vessels of all sizes contributing to the diffuse leakage of blood from the gastric surface. |
TABLE 4.5 Pathology of Acute Erosive Gastritis | |
|---|---|
|
The histologic features depend on the severity and duration of the underlying insult; they are often not as dramatic as the gross features. Features associated with acute gastritis are listed in Table 4.5. Mucosal changes range from hyperemia, surface erosions, and acute inflammation to massive mucosal necrosis (Fig. 4.34), sloughing, and eventual scarring. Lesions seen in biopsies are typically early. More severe changes are usually seen at the time of autopsy. Mild stress gastritis may be difficult to distinguish from biopsy trauma. More severe disease shows extreme vascular congestion with dilation and hemorrhage into the superficial lamina propria (Fig. 4.34) often associated with acute inflammation (Fig. 4.34).
Erosions appear as discrete, superficial oval or circular areas of mucosal necrosis and tissue loss that do not extend deeper than the muscularis mucosae and have sharply defined, often raised edges; edema; and superficial epithelial necrosis (Fig. 4.34). Numerous neutrophils infiltrate the gastric pits and glandular lumina (Fig. 4.35). The inflammation usually spares the deepest glands. True granulation tissue is absent. Rather, the eroded cavity contains an exudate of proteinaceous fluid, debris, neutrophils, and red cells. The mucosa contains superficial fibrin deposits (Fig. 4.34). Chronic inflammation is absent in the acute phase. Only a minimal reparative fibroblastic response occurs when the injury is minor. Healing occurs in days to weeks following removal of the causative factor(s).
The healing phase (Table 4.5) is characterized by proliferation of pluripotential stem cells in the mucous neck region, pit elongation, a pseudostratified or syncytial appearance of the superficial epithelium, and vascular congestion (Fig. 4.35). The stem cells differentiate into foveolar cells above and specialized glandular epithelial cells below, reconstituting a normal mucosal architecture within a few days. Proliferating mucous neck cells contain abundant basophilic cytoplasm, increased mitoses, and an increased nuclear:cytoplasmic ratio, and appear mucin depleted. Despite their potentially alarming cytologic features (Fig. 4.35), the nuclei of the regenerating epithelium retain a basal orientation and contain vesicular chromatin and a prominent solitary eosinophilic nucleolus. The nuclear
pleomorphism and atypical mitoses characteristic of neoplasia are usually absent. Residual clusters of neutrophils may reside within the pits and the surrounding lamina propria may be inflamed. One must be careful not to mistake regenerative changes for carcinoma. Atypical regenerative epithelium still retains a normal glandular architecture. If one can line the glands up parallel with one another in a regular fashion, perpendicular to the mucosal surface, if there is acute inflammation and if lamina propria separates the glands, one should be extremely cautious before making a diagnosis of malignancy, even in the face of extreme epithelial atypia.
pleomorphism and atypical mitoses characteristic of neoplasia are usually absent. Residual clusters of neutrophils may reside within the pits and the surrounding lamina propria may be inflamed. One must be careful not to mistake regenerative changes for carcinoma. Atypical regenerative epithelium still retains a normal glandular architecture. If one can line the glands up parallel with one another in a regular fashion, perpendicular to the mucosal surface, if there is acute inflammation and if lamina propria separates the glands, one should be extremely cautious before making a diagnosis of malignancy, even in the face of extreme epithelial atypia.
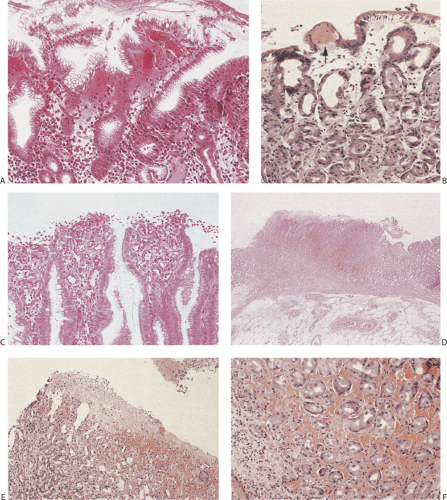 FIG. 4.34. Early changes of acute erosive gastritis. A: The earliest changes consist of vascular dilation in the superficial lamina propria. The overlying epithelium remains intact. B: Small thrombi develop (arrow) and the superficial lamina propria becomes increasingly edematous. C: With further disease progression, the tops of the gastric surface become eroded with loss of the surface epithelium. D: In larger lesions, extensive areas of transmucosal necrosis are present. E: Portions of the superficial epithelium degenerate, forming amorphous pinkish fibrinous debris on the surfaces of the mucosa. The glands become widely dilated and the surrounding lamina propria is infiltrated with extravasated red cells. F: Higher magnification of the extravasated red cells. |
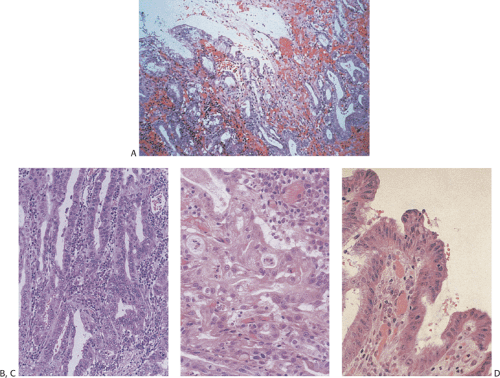 FIG. 4.35. Evolving erosive gastritis. A: The mucosal surface is eroded and as a result mucous neck regions become hyperchromatic and appear regenerative. B: With further disease progression, one sees a marked expansion of the mucous neck region with the regenerative cells showing significant reparative atypia. C: The surface cells acquire a syncytial appearance. D: Eventually, the entire surface becomes re-epithelialized, although the epithelium appears immature and has a large nuclear:cytoplasmic ratio. Some residual syncytial knots remain. |
Superficial erosions usually heal completely, without evidence of scarring, providing the inciting agent disappears. In patients with deeper lesions, complete regeneration of the gastric glands rarely occurs. Rather, mild mucosal scarring results (Fig. 4.36).
Ethanol-Induced Gastritis
The extent of alcohol-induced injury results from the quantity of alcohol ingested as well as its mucosal contact time (67). Injury usually requires gastric alcohol concentrations >10%. The presence of a concurrent HP infection may augment the alcohol-induced injury. Alcohol contacting the superficial gastric mucosa impairs mucus synthesis and secretion and damages epithelial cells, causing them to become necrotic and slough, leaving the underlying mucosa exposed to the alcohol and to gastric luminal acid (68). Acid back-diffusion increases mucosal blood flow, capillary permeability, and acid secretion. Increased capillary permeability leads to interstitial edema. Vasoactive mediator release from mast cells, endothelial cells, and neutrophils triggers venoconstriction and plasma transudation. The neutrophils generate superoxide anion and hyperchlorous acid in a manner similar
to that seen in stress gastritis (69). Arterial and arteriolar dilation rapidly follow, leading to marked congestion, edema, hemorrhage, cellular translocation, ischemia, and cell membrane damage sufficient to cause local edema, hypoxia, hemorrhage, and cellular necrosis (Fig. 4.37). Alcohol penetration into the congested tissues causes hemolysis, vascular congestion, protein precipitation, vascular stasis, thrombus formation, and capillary leakage (70). Neuropeptides stimulated by the alcohol affect blood vessels, leukocytes, and epithelium, and aid in activating inflammatory mediators (71).
to that seen in stress gastritis (69). Arterial and arteriolar dilation rapidly follow, leading to marked congestion, edema, hemorrhage, cellular translocation, ischemia, and cell membrane damage sufficient to cause local edema, hypoxia, hemorrhage, and cellular necrosis (Fig. 4.37). Alcohol penetration into the congested tissues causes hemolysis, vascular congestion, protein precipitation, vascular stasis, thrombus formation, and capillary leakage (70). Neuropeptides stimulated by the alcohol affect blood vessels, leukocytes, and epithelium, and aid in activating inflammatory mediators (71).
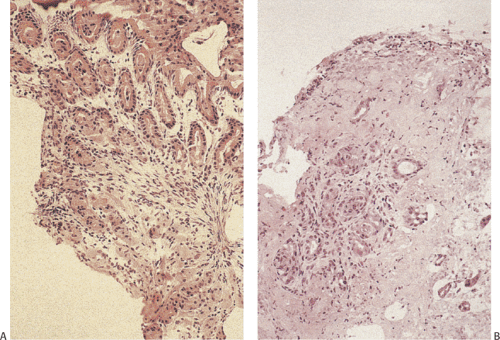 FIG. 4.36. Healed erosion. A: A focal area of fibrosis of the lamina propria distorts the glands. B: This figure shows more extensive lamina propria fibrosis and collagenization with only a few atrophic glands remaining. The surface has not re-epithelialized in this particular specimen, indicating the presence of both acute and more chronic recurrent damage. |
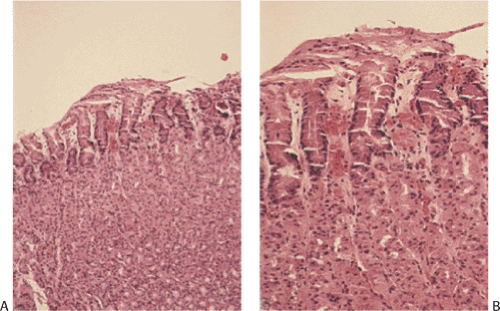 FIG. 4.37. Alcohol-induced gastric injury. A: Low magnification demonstrates the presence of superficial damage in the oxyntic mucosa. B: Higher magnification shows the presence of superficial loss of the epithelium, edema, vascular congestion, and almost no inflammation. |
Actively drinking alcoholics predominantly show multiple areas of subepithelial hemorrhage, prominent mucosal edema in the adjacent nonhemorrhagic mucosa, and only mild inflammation. The edema may be severe enough to extend into the submucosa (72). The foveolar epithelium overlying the lamina propria hemorrhage may appear mucus depleted and show focal loss of nuclear polarity. Tiny erosions may be present in some patients, especially those consuming large quantities of alcohol in a short period of time. These resemble the lesions seen in stress gastritis and predominantly involve the proximal stomach. In these patients there may be focal necrosis of the foveolar epithelium along with focal neutrophilic infiltrates in the gastric pits. Chronic ethanol ingestion increases mucosal expression of EGF and other growth factors (68) leading to increased cell proliferation. Differentiation of cells in the proliferating mucous neck region replaces the damaged cells.
Drug-Induced Gastritis and Gastropathy
Many drugs produce gastric erosions, hemorrhage, and necrosis, the most common of which are NSAIDs and aspirin. The pathogenesis of the injury varies. Some drug-induced and stress-induced ulcers share common pathogenetic events, but the factors that lead to the initial cellular damage may differ. The fact that many drugs produce similar changes whether administered intravenously or orally suggests that mucosal contact need not occur to produce the damage.
Aspirin/Nonsteroidal Anti-Inflammatory Drugs
The nature of aspirin-related injury depends on whether the drug ingestion is acute or chronic. One-time aspirin ingestion causes subepithelial hemorrhages within an hour, and regular intake over a 24-hour period leads to gastric erosions in many individuals. Chronic ingestion often results in less severe damage than acute ingestion because mucosal adaptation occurs, making the mucosa resistant to injury. The adaptive response involves decreased neutrophilic infiltration and extensive epithelial proliferation (73). Aspirin-induced damage results from its toxic effects as well as by decreasing mucosal defenses (74). The physicochemical property of aspirin aids in its rapid absorption, mucosal accumulation, and mucosal barrier breaking effects. Salicylates in aspirin become trapped inside gastric epithelia interfering with ATPase-dependent processes and leading to increased membrane permeability. Eventually osmotic swelling and cell death develop. Additionally, small aspirin fragments may become embedded in the mucosa. These produce circular erosions or ulcers surrounded by hemorrhagic zones. Adjacent erosions become linked by linear mucosal cracks. The aspirin particles then fall into the cracks and become walled off in mucus until they dissolve.
Nonsteroidal Anti-Inflammatory Drugs
NSAIDs are a common cause of grossly visible gastric injury and are responsible for some of the most severe drug injury seen in the United States (75). Gastric injury typically complicates the use of NSAIDs prescribed by physicians. However, consumption of high-dose over-the-counter NSAIDs also causes significant gastrointestinal injury (76). Gastroduodenal lesions develop in 31% to 68% of patients on chronic therapy; up to 25% have gastric ulcers (77). The vast majority of individuals utilizing NSAIDs are 60 years of age or older. These patients are particularly susceptible to develop GI hemorrhage and gastric ulcers, in part due to the fact that mucosal prostaglandin levels decrease in the elderly (78).
NSAIDs cause direct local mucosal toxicity and inhibit hydrogen sulfide generation (79) and cyclooxygenase enzymes. The latter leads to reduced prostaglandin synthesis. As noted earlier, prostaglandins are critical to maintaining the integrity of the mucosal barrier. Most NSAIDs are weak organic acids and in the highly acidic stomach, are un-ionized so that they are lipid soluble and diffuse freely into the epithelium elevating intracellular pH. The damaged mucosa becomes leaky allowing acid back-diffusion, peptic injury, erosions, hemorrhage, and other damage to occur. Coexisting HP infections increase mucosal susceptibility to NSAID-mediated damage and increase the risk of ulceration and bleeding (80).
NSAIDs produce acute mucosal lesions within 7 days of administration by the mechanisms shown in Figure 4.38. Altered blood flow and increased leukocyte–endothelial interactions in the gastric microcirculation occlude microvessels, further reducing mucosal blood flow. The inflammatory cells also release various procoagulants, inflammatory mediators, proteases, and oxygen-free radicals that further damage the endothelium and the underlying connective tissue (81). NSAID effects are summarized in Table 4.6 (74).
NSAIDs cause several types of injury: Acute hemorrhagic gastritis, erosions, chemical gastropathy (discussed in a later section), ulcers, and perforation. Because of the direct local toxicity caused by NSAIDs, the damage is patchy and is increased in areas of mucosal contact. Thus, injury is more common in dependent parts of the stomach (antrum and body along the greater curvature). Gastric ulcers are of greater clinical significance than erosions because of their chronicity and their potential for perforation and significant bleeding.
The acute hemorrhagic gastritis is characterized by a damaged surface epithelium with edema, hemorrhage into the superficial lamina propria, and variable inflammation, resembling stress gastritis. The degree of inflammation tends to be minimal. However, neutrophilic infiltrates are present if erosions or ulcers develop. The hemorrhagic erosions usually heal within a few days. Prominent eosinophilia may also be present, as may increased epithelial apoptosis. Biopsies
taken of antral lesions thought to be erosions by endoscopists are frequently areas of chemical gastropathy with moderate to marked capillary dilation. The epithelium may appear very atypical (Figs. 4.39 and 4.40). The diagnosis of NSAID-induced gastritis can be problematic in the absence of a history of NSAID ingestion, but the presence of changes resembling stress gastritis, or a chemical gastropathy, especially if increased eosinophils are present, may suggest the diagnosis.
taken of antral lesions thought to be erosions by endoscopists are frequently areas of chemical gastropathy with moderate to marked capillary dilation. The epithelium may appear very atypical (Figs. 4.39 and 4.40). The diagnosis of NSAID-induced gastritis can be problematic in the absence of a history of NSAID ingestion, but the presence of changes resembling stress gastritis, or a chemical gastropathy, especially if increased eosinophils are present, may suggest the diagnosis.
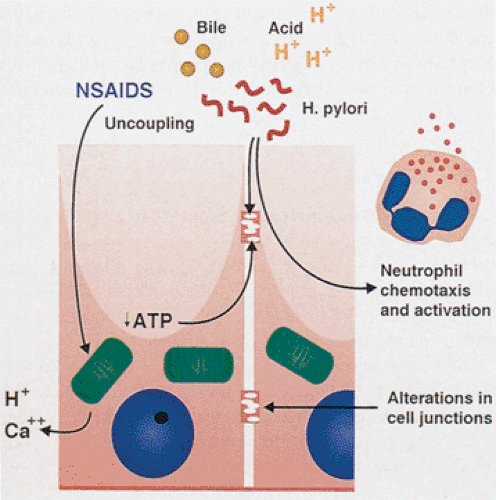 FIG. 4.38. Diagram of the mechanism of nonsteroidal anti-inflammatory drug (NSAID) damage. Following NSAID absorption, there is uncoupling of mitochondrial oxidative phosphorylation leading to reduced adenosine triphosphate (ATP) levels, which in turn result in the loss of intercellular junctional integrity and increased mucosal permeability. Also as a result of the mitochondrial oxidative phosphorylation uncoupling, an efflux of calcium and hydrogen ions from mitochondria occurs, further depleting ATP stores and promoting oxygen radical damage. The damaged cell releases arachidonic acid, but the conversion of arachidonic acid to prostaglandins is prevented by the NSAID inhibition of cyclooxygenase. As a result, the damage of the gastric mucosa is more prolonged than would ordinarily be the case. As a result of the damage, the mucosa becomes vulnerable to luminal aggressive factors, which include acid, pepsin, bile, and Helicobacter pylori. |
Proton Pump Inhibitors
Proton pump inhibitor (PPI) therapy may induce changes in G cells, ECL cells, and parietal cells. Mild G-cell and ECL-cell hyperplasias (but not carcinoid tumors) occur and may be diffuse, linear, or micronodular (82). The micronodular hyperplasia is more likely to occur in patients with HP infections. Parietal cell changes include increases in cell size and number, leading to swelling (Fig. 4.41) and convex bulging of the apical cell membranes into the glandular lumens, which imparts a serrated appearance to the normally round or tubular glandular lumens (83). This change affects over 90% of patients treated for 1 year with daily doses of 20 to 40 mg of omeprazole. The parietal cell protrusions appear to be related to the hypergastrinemia (83,84). For this reason, parietal cell protrusions are not specific for PPI therapy. They can also be seen in patients with HP gastritis, gastric ulcers, morbid obesity, Zollinger-Ellison syndrome, or gastric cancer (83).
TABLE 4.6 Nonsteroidal Anti-inflammatory Drug Effects | |
|---|---|
|
Patients on long-term treatment may develop tiny fundic gland polyp (FGP)-like lesions. They contain glandular cysts measuring between 0.25 and 7 mm in diameter (Fig. 4.41). These are lined by flattened parietal and chief cells; they may also contain foveolar cells. The lesions may disappear after PPI therapy is withdrawn and reappear after the reintroduction of the therapy (85). While there appears to be a relationship between FGPs and PPIs, the data are inconsistent and not all patients develop the polyps. Additionally, some PPI-associated fundic gland cysts lack superficial foveolar dilation, a hallmark of FGPs. PPIs also slightly increase the number of apoptotic bodies in the antral mucosa (86).
Steroids
The association between peptic ulcer disease and corticosteroids has been debated for years. There appears to be a small increased risk of gastric ulceration and bleeding in steroid users; the concomitant use of steroids and NSAIDs associates with an up to 10-fold increased risk of an upper GI bleed. Steroids stimulate G-cell hyperplasia (87), indirectly increasing acid production by parietal cells. They also decrease epithelial turnover and mucin secretion, thereby impairing mucosal barrier protection. There may be acute inflammatory cells in the lamina propria and apoptotic debris in the glandular lumens (Fig. 4.42).
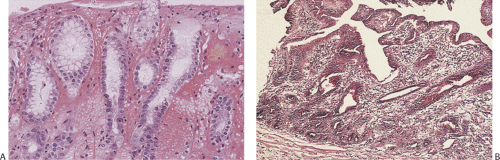 FIG. 4.39. Nonsteroidal anti-inflammatory drug (NSAID) damage. A: A gastric biopsy from the body region of a patient on NSAIDs. The patient showed endoscopic evidence of multiple petechial hemorrhages. The biopsy shows that the changes mainly affect the superficial mucosa with edema, telangiectasia, and little or no inflammatory infiltrate. B: The mucosa appears regenerative with loss of glands and irregularity of the remaining pits. |
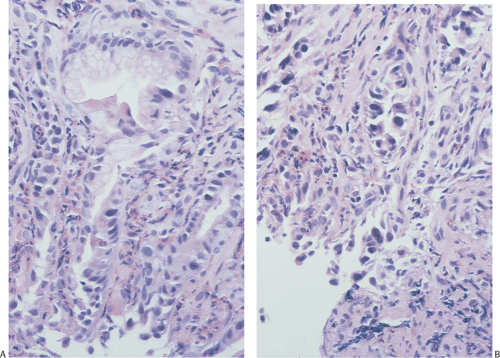 FIG. 4.40. Nonsteroidal anti-inflammatory drug (NSAID) injury. A: The regenerative changes that develop following NSAID-induced injury can be cytologically alarming. The atypia primarily affects the cells in the mucous neck region. A clue to their regenerative, nonneoplastic nature is evidence of maturation in the foveolar cells. B: When the biopsy is fragmented without the ability to see the foveolar cell maturation, the diagnosis is more difficult. |
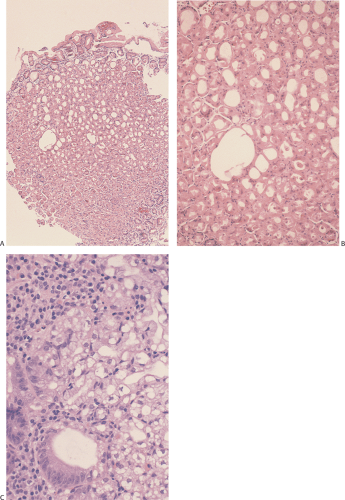 FIG. 4.41. Proton pump inhibitor–induced injury. A: Cystic changes in the mucosa. B: Higher magnification. C: Vacuolization of the parietal cells. |
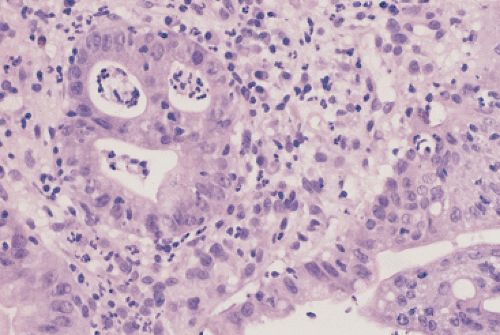 FIG. 4.42. Patient with aplastic anemia treated with steroids. There is acute inflammation in the lamina propria and the gland, and the glands contain apoptotic debris. |
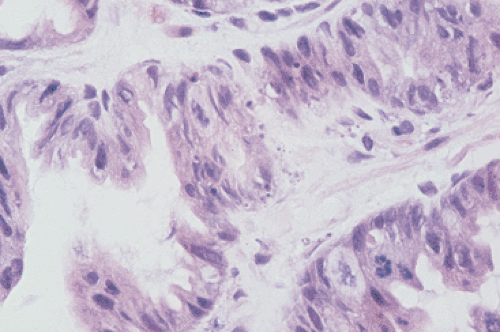 FIG. 4.43. Chemotherapy-associated gastric changes showing increased mitotic activity and enlarged atypical cells. The nuclear:cytoplasmic ratio is not increased. |
Chemotherapy
Patients undergoing chemotherapy develop many complications due to direct mucosal injury, vomiting (leading to potential esophagogastric tears), drug-induced immunosuppression, and thrombocytopenia. Cisplatin, doxorubicin, nitrosoureas, and vincristine are notorious for inducing nausea and vomiting. Chemotherapy-induced immunosuppression can lead to multiple gastric infections. Hepatic arterial infusion chemotherapy (HAIC) can produce gastric ulcers and alarming epithelial regenerative atypias easily confused with carcinoma (88,89) because of the presence of binucleated or multinucleated cells containing massive nuclei (Fig. 4.43). Features of the atypia of HAIC and neoplasia are compared in Table 4.7. The presence of ring mitoses, which are not characteristic of neoplasias, suggests drug injury. Floxuridine causes epithelial necrosis with the inflammation and epithelial regeneration (90). The epithelial cytoplasm becomes vacuolated and foamy. Other drugs causing severe epithelial atypia include combinations of 5-fluorouracil (5-FU) with either leucovorin or mitomycin. These drug-induced atypias become most difficult to accurately diagnose in superficial gastric mucosal brushing specimens. Vinorelbine predisposes to bezoar formation (91), probably due to toxic myenteric plexus damage.
TABLE 4.7 Comparison of Hepatic Arterial Infusion Chemotherapy (HAIC) and Neoplasia | |||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Iron Pill Gastritis
Acute gastritis develops in individuals consuming large amounts of oral iron. Iron supplements may cause erosions, ulcers, an almost infarctlike gastric mucosal necrosis, and strictures as the ulcers heal. Grossly, grayish or bluish mucosal patches may be present. Superficial edema, inflammation, and necrosis characterize the histologic changes. A layer of brownish pigment that stains positively with iron stains (Fig. 4.44) may cover the epithelial surface and extend into the superficial gastric pits. Some of the pigment may appear crystalline. The majority of the iron that is present is in an extracellular location. Iron pigment may lie in granulation tissue, on the top of damaged epithelium, in the glands or lamina propria, or in the submucosa (Fig. 4.45) (92). Focal epithelial or stromal cell deposition is less common than deposits in the lamina propria and surface unless the
patient has also had transfusions or has alcoholic liver disease. Features of chemical gastropathy may also be present (93). The changes differ from those seen in hemosiderosis in that patients with hemosiderosis or hemochromatosis lack significant inflammation (unless there is a coexisting gastritis) and the iron predominantly localizes within cells, especially the epithelium of deeper glands. Erosions are not present.
patient has also had transfusions or has alcoholic liver disease. Features of chemical gastropathy may also be present (93). The changes differ from those seen in hemosiderosis in that patients with hemosiderosis or hemochromatosis lack significant inflammation (unless there is a coexisting gastritis) and the iron predominantly localizes within cells, especially the epithelium of deeper glands. Erosions are not present.
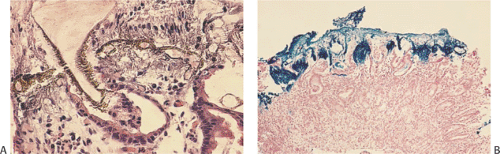 FIG. 4.44. Changes associated with high-dose iron ingestion. A: Note the superficial damage and edema of the lamina propria. A layer of iron is deposited on the surface of the gastric mucosa as well as extending into the upper portions of the gastric pits. B: Prussian blue stain of the same biopsy at a lower magnification showing the prominent coating of the gastric mucosa with iron and extension into the pits. |
Prostaglandin Therapy
Prostaglandin E infusions, used to treat congenital heart disease and other neonatal and adult disorders, induce gastric outlet obstruction secondary to foveolar hyperplasia and elongation of the gastric pits that results in marked antral hyperplasia (94).
Cocaine Use
Cocaine abuse can cause gastric injury with the newer forms being more toxic than cocaine hydrochloride (95). Affected patients tend to be young males. Cocaine causes intense vasoconstriction, focal ischemia, and perforation (95,96). Granulomas may be present, presumably due to cutting the drugs with foreign materials (Fig. 4.46).
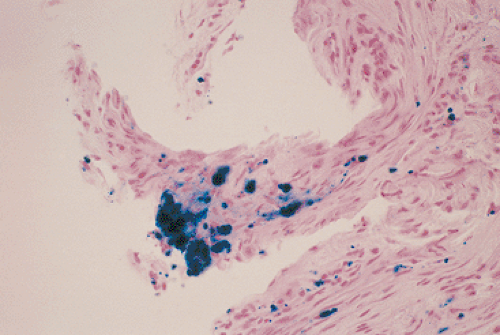 FIG. 4.45. Iron-pill gastritis. Note the prominent iron deposits in the submucosa. |
Gastric Mucosal Calcinosis
Rare patients have crystalline deposits in the superficial gastric mucosa that consist of aluminum, phosphorus, calcium, and chlorine. They occur in transplant patients on long-term aluminum-containing antacid therapy or sucralfate therapy (97). The deposits lie just below the foveolar epithelium, sometimes rimmed by macrophages. They show variable degrees of calcification and often appear refractile (Fig. 4.47). There may be coexisting gastritis.
Other Drugs
Kayexalate resin crystals can be present in the stomach in patients treated for hyperkalemia. Sodium cations are released from the resin and exchanged for hydrogen ions in the gastric acid milieu. As the resin passes through the intestines, hydrogen is exchanged for potassium, which is then eliminated in the feces along with the remainder of the altered resin, thereby lowering serum potassium levels. The crystals have a characteristic refractile crystalline mosaic pattern that resembles fish scales that distinguishes Kayexalate crystals from cholestyramine crystals, which they resemble (98,99). This pattern is faintly seen in hematoxylin and eosin-stained sections and is better demonstrated with acid fast, Alcian blue, or Diff-Quik stains. The crystals do not polarize. The crystals are always located in a luminal location, either adherent to intact surface epithelium or admixed with inflammatory exudates in patients with ulcers or erosions (98,99). In some cases, Kayexalate crystals aggregate with mucous secretions and clotted blood to form gastric bezoars.
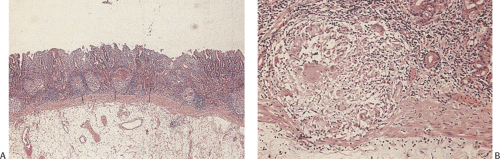 FIG. 4.46. Granulomatous gastritis in a cocaine addict. A: Numerous granulomas lie in the basal portion of the mucosa just above the muscularis mucosae. They are surrounded by a prominent mononuclear cell infiltrate. B: Higher magnification of one of these granulomas shows the basal location and the presence of a granulomatous response containing giant cells. |
Some patients on colchicine develop gastritis or antral erosions. The most discriminating feature indicating colchicine damage is the presence of abundant epithelial mitotic figures in metaphase arrest (100), particularly in the proliferative zone in the mucous neck region. Metaphase mitoses appear as enlarged epithelial cells with condensed chromatin in a ring formation in the center of the cell (ring mitoses). These changes are accompanied by marked foveolar cell hyperplasia resulting in crowded, enlarged, distorted epithelial cells; epithelial pseudostratification; and loss of cellular polarity. However, in contrast to the large size of the cells, the nuclei are small, hyperchromatic, and compressed to the cellular periphery. The changes are more prevalent in the gastric antrum than in the gastric body (100).
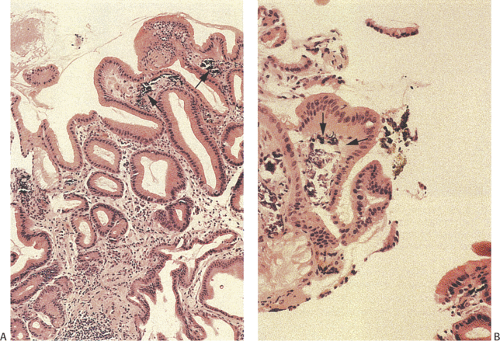 FIG. 4.47. Mucosal calcinosis. A: Low-magnification photograph of the gastric mucosa showing the presence of superficial aggregates of deeply pigmented crystalline material (arrows) that were positive with Von Kossa stains. B: Higher magnification showing the details of these mucosal deposits (arrows). |
Ticlopidine sometimes causes lymphocytic gastritis, and methyldopa treatment sometimes leads to the development of autoimmune gastritis (101). Both interleukin-4 (IL-4) and tumor necrosis factor (TNF), agents used to treat patients with advanced cancer, cause acute gastric mucosal injury and hemorrhagic necrosis (102). Some drugs alter gastric motility, including anticholinergics, adrenergic agents, dopaminergic agents, narcotics, and erythromycin.
Acute Corrosive Gastritis
Extensive acute gastritis results from ingestion of corrosive agents (Fig. 4.48), be it accidental or suicidal. The types of agents that cause injury resemble those damaging the esophagus (see Chapter 2). Ingestion causes rapid and widespread gastric mucosal necrosis. The mucosal surface may appear black due to digested blood and mucosal necrosis. The damage often localizes to the prepyloric region, due to the stasis that occurs there allowing longer mucosal contact times with injurious agents than would otherwise exist. However, if only small quantities of the corrosive agent are consumed, then the injury may be more proximal in location. Initially there is mucosal hemorrhage and edema. The injury may extend deep into the gastric wall and patients with myonecrosis may perforate. Severe acute changes consist of coagulative necrosis (Fig. 4.49). The damage is patchy, with the most severe damage occurring in the areas of mucosal contact. Alkalis usually injure the esophagus more severely than the stomach; the reverse occurs with acids.
If the patient survives the acute event, fibrosis causes progressive stricturing. If the process progresses, complete obstruction may result. Perforation, peritonitis, or massive hemorrhage complicates corrosive gastritis. One classifies corrosive gastritis into three grades, in a manner similar to that used for corrosive esophageal burns (Table 2.10).
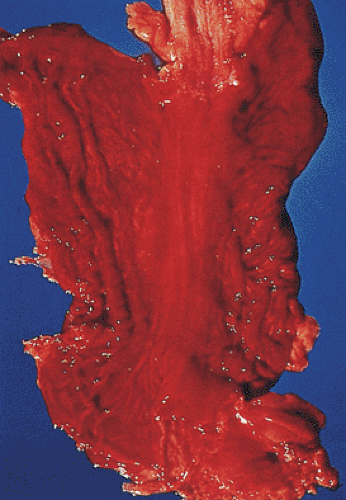 FIG. 4.48. Corrosive gastritis from a suicidal ingestion of acid. |
Ischemic Gastritis
While focal ischemia mediates many of the changes seen in acute (stress) gastritis, gastric infarction is rare because of the rich gastric vasculature. Severe, diffuse arterial sclerosis coupled with smoking and systemic hypertension (103) can lead to severe gastric damage and even gastric infarction (Fig. 4.50). Less severe ischemia develops secondary to vasculitis, bacterial gastritis, previous gastric surgery, disseminated intravascular coagulation (DIC), or volvulus (103). The features of ischemic gastritis include a coagulative
necrosis (Fig. 4.50), the extent of which reflects the degree and duration of the ischemia. Transmural infarction is a rare and usually terminal event. The histologic features resemble those seen in the intestines and are discussed more extensively in Chapter 6.
necrosis (Fig. 4.50), the extent of which reflects the degree and duration of the ischemia. Transmural infarction is a rare and usually terminal event. The histologic features resemble those seen in the intestines and are discussed more extensively in Chapter 6.
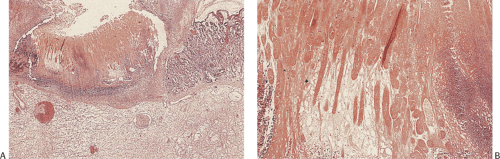 FIG. 4.49. Lye ingestion in suicide attempt. A: Much of the stomach had become completely necrotic. The lesions were focally distributed throughout the gastric mucosa as seen in A. B: Higher magnification demonstrating the coagulative necrosis and edema of the gastric mucosa. |
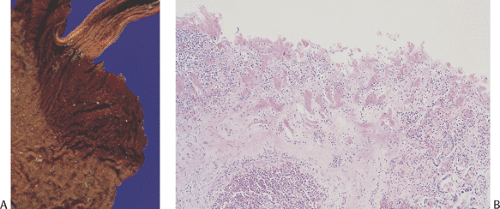 FIG. 4.50. Ischemic necrosis of the cardia and upper part of the fundus. A: This patient died of severe cardiovascular collapse. The patient was a known smoker, was hypertensive, and had previously undergone a surgical procedure. The death occurred during the postoperative period. B: Coagulative necrosis is seen affecting the gastric glands. |
Radiation Gastritis
Patients receiving radiation in doses in excess of 4,000 rads demonstrate both acute and chronic gastritis with areas of epithelial necrosis and shallow ulcers (104). Gastric radiation injury assumes one of three forms:
Acute radiation gastritis develops a few days to a few months following the radiation exposure. Inflammation is present early but it eventually diminishes. Extensive mucosal necrosis and superficial ulcers may be present. The endothelium swells, reducing vascular luminal caliber. Submucosal capillary ectasia is present. As the mucosa heals, it regenerates, but variable degrees of atrophy, fibrosis, edema, and endarteritis persist. The cytologic features may resemble those induced by many chemotherapeutic agents. Indeed, many cancer patients show the effects of combined chemoradiation.
One to two months following radiation exposure, some patients develop a deep acute ulcer, which usually walls off before perforation occurs. The ulcer represents the long-term effects of vascular damage and ischemia. Stigmata of radiation damage are histologically evident as evidenced by the vascular alterations and the presence of radiation fibroblasts in the surrounding fibrotic stroma (104).
A month to several years following radiation exposure, a chronic ulcer, indistinguishable from a peptic ulcer, may develop. The only clues to its etiology are the clinical history, an unusually prominent antral fibrosis with obliterative endarteritis or foamy macrophages involving the submucosal blood vessels, and the presence of atypical hyperchromatic fibroblasts (“radiation fibroblasts”). The submucosa develops capillary telangiectasia and arterial wall hyalinization with intimal fibrosis. Patients may also develop severe atrophic gastritis. Even though mucosal regeneration occurs, gastric acid output remains reduced; many patients develop hypochlorhydria.
Infectious Gastritis
The stomach normally contains <103 organisms/mL, with most coming from the oral cavity. Gastric pH determines the gastric bacterial content; pH levels below 4.0 kill most bacteria. If the pH is not at or below this level, the stomach does not sterilize itself and an abnormal flora may become established. Normal gastric motility and emptying also protect against bacterial infections. When dysmotility or pyloric obstruction develops, anaerobic organisms may accumulate. Not uncommonly one finds oral bacteria lying in the gastric lumen. They have a typical diplococcal or tetrad morphology and reach the stomach when swallowed with other oral contents or when they are carried there via the endoscope. These organisms have no clinical significance.
Helicobacter Pylori
Transmission
HP infections occur worldwide, although there are marked geographic variations in their incidence. The prevalence of HP infections in adults ranges from <15% in some populations to virtually 100% in less well-developed areas. There are also substantial variations among different ethnic groups in the same geographic locale (105). In developing countries, people become infected much earlier in life than in
developed countries. The prevalence of HP infections correlates with lower socioeconomic status (106). The human stomach is the primary reservoir for the organism and it is transmitted via an oral–oral route and possibly via a gastric–oral and fecal–oral route (107). The infection is easily passed from one family member to another, particularly in areas of dense housing. Children under the age of 5 are most susceptible to HP infections. HP infections are present in gastric biopsies of 16.8% to 55% of children with abdominal pain, upper gastrointestinal symptoms, and histologic evidence of acute and/or chronic gastritis (106). The infection prevalence in developing countries can be as high as 75% by age 25. There has been a recent decline in the incidence of HP infections in the developed world, largely due to improved living conditions and a decrease in living density and family size. Even though the prevalence of HP infections is currently decreasing, at least 50% of the world population is infected with the organism (108).
developed countries. The prevalence of HP infections correlates with lower socioeconomic status (106). The human stomach is the primary reservoir for the organism and it is transmitted via an oral–oral route and possibly via a gastric–oral and fecal–oral route (107). The infection is easily passed from one family member to another, particularly in areas of dense housing. Children under the age of 5 are most susceptible to HP infections. HP infections are present in gastric biopsies of 16.8% to 55% of children with abdominal pain, upper gastrointestinal symptoms, and histologic evidence of acute and/or chronic gastritis (106). The infection prevalence in developing countries can be as high as 75% by age 25. There has been a recent decline in the incidence of HP infections in the developed world, largely due to improved living conditions and a decrease in living density and family size. Even though the prevalence of HP infections is currently decreasing, at least 50% of the world population is infected with the organism (108).
Recurrent infections are usually a persistent infection rather than the acquisition of new infections. Infection with more than one HP strain can also occur (109). When AIDS patients become infected, the disease may exhibit particularly virulent characteristics and large numbers of organisms may be present.
Pathogenesis
HP is highly adapted to occupy a special ecologic gastric niche with unique features that allow it to enter the mucus of the mucosal barrier, attach to the epithelium, evade immune responses, proliferate, and colonize the gastric mucosa. The eventual outcome of HP infections reflects strain-specific, environmental, and host-related factors. After they are ingested, HP organisms must evade the bactericidal activity of the gastric lumen and enter the mucous layer. Corkscrewlike bacterial movement and enzyme production (particularly urease and lipase) are important early in the infection (110,111). Bacterial proteases digest gastric mucin (194,195,196,197) facilitating bacterial movement and urease protects the HP from the luminal acid by creating an alkaline microenvironment around the bacterium (110,111).
HP bacteria normally reside in the unstirred layer of gastric mucus. They wind down to the epithelial surface, moving easily through the viscous environment above it, to attach themselves to the apical membranes of foveolar cells (Fig. 4.51). Bacterial adhesins recognize cell surface–specific proteins facilitating epithelial colonization. The best characterized adhesin, BabA, is a 78-kD outer bacterial membrane protein that binds to the fucosylated Lewis B blood group antigen (112). The Lewis blood group terminal carbohydrate structures are present on the ends of MUC1 carbohydrate side chains as well as on secreted mucins. MUC1 is highly polymorphic and evidence suggests that functional allelic differences affect infection susceptibility (113). HP bacteria also bind to MUC5AC, a major mucin produced by foveolar cells (114). The specificity of the binding, together with the limited distribution of the receptors, results in a restricted range of HP-colonized tissues. Bacteria unable to adhere to the epithelium are rapidly cleared from the mucosa. HP bacteria preferentially attach at or near intercellular junctions, penetrating the junctional complexes moving down along the lateral cell membranes. This disrupts intercellular tight junctions between viable cells, allowing luminal contents, including acid, to flow between the cells.
 FIG. 4.51. Helicobacter pylori gastritis. Diff-Quik–stained section demonstrating spirally shaped bacteria intimately adherent to the surfaces of the epithelium as well as lying in the intercellular spaces. Both coccoid and spiral forms are present at the free surface. |
Most HP strains secrete the vacuolating cytotoxin VacA (115). The toxin inserts itself into the epithelial cell membrane and forms a hexameric anion-selective, voltage-dependent channel through which bicarbonate and organic anions can be released, possibly providing HP bacteria with their nutrients (116). VacA (117) also inhibits T-lymphocyte activation (118). Certain vacA gene variants produce more severe disease than others (119).
Most HP bacteria possess the cag pathogenicity island (cag PAI) that contains 29 distinct genes (120). Some of these genes facilitate translocation of the CagA protein into foveolar cells (121). Once in the cells, CagA is phosphorylated and binds to the SHP-2 tyrosine phosphatase (122), leading to host growth factor–like cellular responses, cytokine production, and cell proliferation (123). These cytokines mobilize leukocytes to areas of immune challenge. An EPIYA-repeat polymorphism influences the magnitude and duration of phosphorylation-dependent CagA activity (124). CagA also plays a major role in disruption of the apical junctional complexes (125). cagA-positive strains associate with increased epithelial cell apoptosis (126).
The oipA (outer inflammatory protein) gene encodes one of the outer membrane proteins and is an inflammation-related gene near, but not in, the CagA PAI. oipA functional status correlates with clinical presentation, HP density, and gastric inflammation. Cag PAI, babA2, and vacA status may
be a surrogate marker for a functional oipA gene (127). OipA and the cag PAI are both necessary for full activation of the IL-8 promoter (128). Nitric oxidase synthase and cyclooxygenase-2 are induced by HP infections; these enzymes modulate the inflammatory responses.
be a surrogate marker for a functional oipA gene (127). OipA and the cag PAI are both necessary for full activation of the IL-8 promoter (128). Nitric oxidase synthase and cyclooxygenase-2 are induced by HP infections; these enzymes modulate the inflammatory responses.
Host Responses to Helicobacter pylori
HP infections cause gastric inflammation (gastritis) in almost all infected persons, although the severity of the changes varies among individuals. The injury results from both the infection and its associated inflammation. Bile reflux and dietary irritants may further enhance the deleterious bacterial effects. Additionally, anti-HP antibodies that cross-react with the gastric mucosa induce further damage (129). Some HP-infected patients develop an autoantibody response directed at the H+-K+-ATPase pump in parietal cells leading to atrophy of the corpus (129).
Initially neutrophils are recruited to the infected site, followed by the recruitment of T and B lymphocytes, plasma cells, and macrophages. The neutrophilic infiltrate and mononuclear phagocytic activation may be facilitated by bacterial urease production and induction of nitric oxide synthase and cyclooxygenase (130). HP infections generate significant cellular and humoral responses via antigenic stimulation of mucosal monocytes and T cells. The inflammatory cells produce numerous cytokines (TNF-α, interferon-γ, and interleukins 1, 6, and 8), prostaglandins, proteases, and reactive oxygen metabolites that cause epithelial necrosis and mucosal injury. IL-8, a potent neutrophil-activating chemokine expressed by gastric epithelium, plays a central role in the inflammatory response (127,128). HP bacteria containing the cag PAI induce a stronger IL-8 response than cag-negative strains (131). Some cytokines promote leukocyte adhesion to endothelial cells and others recruit additional leukocytes to the infected site. Mediators of local humoral responses, such as mucosal IgA, attract eosinophils, which then degranulate. Stimulated B cells differentiate into IgM, IgA, and IgG antibody–producing cells (132). IgA promotes complement-dependent phagocytosis and HP killing by polymorphonuclear neutrophils (PMNs). Secretory IgA synergizes with IgG to promote antibody-dependent cell-mediated cytotoxicity induced by PMNs, monocytes, and lymphocytes. High anti-HP IgG antibody levels correlate with severe antral gastritis and dense HP antral colonization (132).
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


