The Neoplastic Stomach
Gastric neoplasms constitute a heterogeneous group of tumors; most are adenocarcinomas. Less common gastric cancers, such as lymphomas, neuroendocrine tumors, and stromal tumors, are discussed in Chapters 17, 18, and 19.
Adenocarcinoma Distal to the Cardia and its Anatomic Precursors
The stomach is divided into three subsites: The cardia, the corpus, and the pyloric antrum. Cancers of the cardia and Barrett-associated adenocarcinomas of the esophagus share similar backgrounds, temporal trends, molecular profiles, and behavioral patterns. These two cancers constitute gastroesophageal junction (GEJ) cancer and are discussed in Chapter 3. The epidemiologic backgrounds and temporal trends of cancers arising distal to the cardia are distinctly different from those arising in the cardia. Noncardia stomach cancer was once the most common human cancer worldwide, but it has shown a steep decline in frequency in the United States and Western Europe, where it is now less common than cancer of the GEJ in white males (1). Distal stomach cancer is still very common in developing countries and among migrants from them.
Epidemiology
Gastric carcinoma shows striking temporal and geographic variations in frequency (2), with a more than 10-fold difference in incidence between countries with high and low rates (Table 5.1). England and Wales offer a good example of the temporal decline in noncardia gastric cancer incidence, with male rates falling from 8.5 to 4.1 between 1971 and 1998, and female rates falling from 3.8 to 1.7 over the same time period (3). First-generation migrants from high-risk countries to low-risk countries continue to experience the high rates of their motherland, whereas their children and grandchildren show rates that approach those of the host country (4). This supports the concept that environmental exposures in early life generate cancer precursors that persist into adulthood and are not reversed by a more favorable environment.
The age-specific gastric cancer incidence rates rise steeply after 50 years of age in all populations, peaking at 673 per 100,000 at age 80 in Miyagi, Japan, compared to 103 per 100,000 for 80-year-old white males in low-risk Los Angeles (2). The wide incidence differences in high- and low-risk populations is already apparent at age 25 when the Miyagi and Korean male rates stand at 2.1 and 4.1, respectively, compared to the Los Angeles white male rate of 0.3 per 100,000. This supports the concept that exposure to environmental hazards begins early in life among people in high-risk populations. Gastric cancer incidence rates for men are approximately twice those of women in all populations for persons over 50 years of age, but the male:female incidence ratio is 1 or less at younger ages. The age-related variation in the male:female incidence ratio suggests that host factors may influence the level of risk of acquiring this cancer.
The rapid decline in the frequency of gastric cancer in the United States since World War II is best appreciated by following the age-adjusted mortality rates for this tumor in white males: 16.34 per 100,000 for the years 1950–1969 and 7.33 per 100,000 for the years 1970–1994 (5). A decrease in gastric cancer incidence since 1973 has occurred in both high- and low-risk countries as is shown in Table 5.2 (2). The earlier decrease in stomach cancer in Western countries was unplanned, resulting from modern refrigeration of foods, year-round access to fresh fruits and vegetables, and, more recently, the treatment of Helicobacter pylori infections.
Gastric cancer arising distal to the cardia does not behave as a single disease, but as several diseases that affect different parts of the stomach and vary in their histologic presentations. Lauren (6) first recognized that distal stomach cancers occur in two forms that differ in their structure and behavior: An intestinal type that consists of intestinal-type glands and a diffuse type that consists of discohesive cells supported by a desmoplastic stroma. The intestinal type is more likely to show vascular spread and hepatic metastases. The diffuse form has a less favorable prognosis, is less likely to have hepatic metastases, and is prone to transperitoneal spread. Both types have decreased in frequency in developed countries and share similar environmental exposures. Age, sex, and blood type, as well as inherited predispositions, determine whether a patient will develop an intestinal or diffuse cancer. The distinction between intestinal and diffuse types of gastric cancer has proven to be valuable in epidemiologic studies and in assessing tumor prognosis. Mixed forms of
these cancers occur, as well as distinctive subtypes of these tumors, but the practical advantages of this simple classification have held up over time.
these cancers occur, as well as distinctive subtypes of these tumors, but the practical advantages of this simple classification have held up over time.
TABLE 5.1 Geographic Variation, Male, Age-standardized Gastric Cancer Incidence Rates per 100,000 | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Both types of gastric cancer occur in high-risk populations, but the proportion of diffuse tumors is increased in low-risk groups. A retrospective analysis of gastric cancer in the SEER (Surveillance, Epidemiology, and End Results) registries of the United States found that diffuse cancers have increased in frequency between the years 1973 and 2000, while intestinal-type cancers have decreased over the same time period (7). A similar trend may account for a recent decrease in antral carcinomas relative to those in the middle third of the stomach in Japan (8), since the proportion of diffuse cancers is higher in the more proximal stomach. The typical differences in intestinal and diffuse gastric cancers are summarized in Table 5.3.
TABLE 5.2 Temporal Trends in Gastric Cancer Incidence Rates, Selected National Registries | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Predisposing Factors and Conditions
The persons at highest risk of acquiring gastric cancer are concentrated in the lower economic strata of developing countries. The initial steps in cancer induction occur in childhood with infection by H. pylori (9). The youngest children of large families living in crowded, unsanitary conditions are at highest risk (10). Familial clusters of stomach cancer result from a shared exposure to environmental hazards (11) and to inherited factors (12).
Genetic Factors
There are a number of types of genetic and epigenetic changes that play a role in the predisposition to or the development of gastric carcinoma (Table 5.4). These include activation of oncogenes, growth factors and growth factor receptors, inactivation of tumor suppressor genes, DNA repair genes, and cell adhesion molecules, as well as alterations in cell cycle regulatory genes. Inherited factors interact with environmental hazards to increase gastric cancer risk in two ways: (a) germline mutations account for well-defined, but infrequent, familial cancer syndromes; and (b) polymorphisms in genes that govern cell cycling or enzymes that catalyze carcinogen detoxification may also influence gastric cancer risk. Other types of changes occur in sporadic tumors.
Inherited gastric cancer predisposition syndromes account for approximately 10% of gastric cancers. The genetic events are known for some but not all of the predispositions. The hereditary nonpolyposis colon cancer syndrome (HNPCC) is an example of the interaction of environmental hazards with germline mutations. Persons with this autosomal dominant condition are at increased risk of developing
cancer at sites other than the colon, including the stomach. HNPCC is due to a germline mutation in mismatch repair genes. The risk of gastric cancer in Korean patients who carry these mutations is 3.2-fold greater than the general population of Korea, where gastric cancer accounts for 25% of all male cancers (12). In occidental countries HNPCC-associated gastric cancer has decreased in frequency in parallel with the decline in sporadic cancer (13), as might be expected if a DNA repair defect is not challenged by genotoxic hazards. The Finnish HNPCC registry has identified 45 patients with gastric cancer in 51 HNPCC families (14), and gastric cancer was the only cancer found in 22 of these. The mean age of gastric cancer development was 6 years older than colon cancer in these HNPCC families. Like sporadic stomach cancer, but unlike most Finnish familial stomach cancers, the HNPCC cancers were intestinal in type and arose in the distal stomach.
cancer at sites other than the colon, including the stomach. HNPCC is due to a germline mutation in mismatch repair genes. The risk of gastric cancer in Korean patients who carry these mutations is 3.2-fold greater than the general population of Korea, where gastric cancer accounts for 25% of all male cancers (12). In occidental countries HNPCC-associated gastric cancer has decreased in frequency in parallel with the decline in sporadic cancer (13), as might be expected if a DNA repair defect is not challenged by genotoxic hazards. The Finnish HNPCC registry has identified 45 patients with gastric cancer in 51 HNPCC families (14), and gastric cancer was the only cancer found in 22 of these. The mean age of gastric cancer development was 6 years older than colon cancer in these HNPCC families. Like sporadic stomach cancer, but unlike most Finnish familial stomach cancers, the HNPCC cancers were intestinal in type and arose in the distal stomach.
TABLE 5.3 Intestinal Versus Diffuse Gastric Cancers | |||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Familial adenomatous polyposis (FAP) may involve the stomach as well as the large bowel. As in the colon, gastric cancers are preceded by the development of dysplasia. The dysplasia may be flat or in polypoid lesions including adenomas, hyperplastic polyps, and fundic gland polyps. An environmental influence is suggested by the observation that Japanese patients with FAP have more frequent gastric adenomas and cancers than westerners with FAP (15,16). This may reflect the higher Japanese exposure to gastric cancer risk factors.
Germline mutations in the E-cadherin/CDH1 gene were first recognized as a source of familial stomach cancer in a New Zealand Maori family (17), and have since been shown to have a worldwide distribution (18,19). This form of familial cancer is now termed hereditary diffuse gastric cancer (HDGC). E-cadherin is a cell adhesion protein. The loss of its function (and loss of its normal expression on the cell membrane; Fig. 5.1) due to gene mutations results in the discohesive cells that characterize sporadic diffuse gastric cancers and lobular breast cancers. It is not surprising, therefore, that familial gastric cancers associated with germline mutations in this gene are diffuse in type and that HDGC kindreds also show an increased frequency of lobular breast cancer (20). Asymptomatic patients who carry the HDGC mutation are treated with prophylactic gastrectomy since the early lesions are undetectable at the time of endoscopy (21). Multiple superficial mucosal cancers may be found at the time of gastrectomy (Fig. 5.1). The chance discovery of similar lesions in an endoscopic biopsy should alert the pathologist to a possible HDGC mutation (22). Lynch et al (13) suggest that these superficial neoplastic foci represent a widespread field effect due to promoter hypermethylation and that dysregulation of genes required for invasion and metastases are much less common.
TABLE 5.4 Types of Genetic Alterations in Gastric Carcinogenesis | |
|---|---|
|
Carriers of BRCA1/2 mutations have a substantial increase in the lifetime risk of breast and ovarian cancer. These mutations have been related to a variety of other sites as well, including the stomach. The Swedish Family Cancer Database assessed the cancer incidence in classified family members (n = 944,723) and compared their cancer experience with that of the general population of Sweden and found a twofold increase in the risk of acquiring stomach cancer before age 70 among male members of families with breast and ovarian cancer (23).
Germline p53 mutations are inherited among families diagnosed with the Li-Fraumeni syndrome. Cancers arise at diverse sites in these families (24). Interaction with environmental factors is suggested by the more common prevalence of stomach cancer in Japanese families with this syndrome than among affected American families (25). Patients with the Peutz-Jeghers syndrome are at very high relative and absolute risk for gastrointestinal and other cancers. The relative risk of stomach cancer risk with this syndrome is 213
(95% confidence interval [CI] = 96 to 368) (26). A mucoid gastric adenocarcinoma associated with germline mutation in the STK11 gene has been identified in a Japanese Peutz-Jeghers family (27).
(95% confidence interval [CI] = 96 to 368) (26). A mucoid gastric adenocarcinoma associated with germline mutation in the STK11 gene has been identified in a Japanese Peutz-Jeghers family (27).
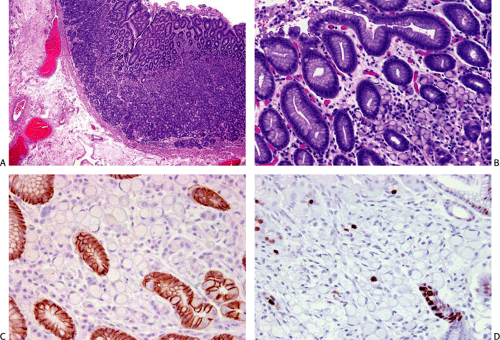 FIG. 5.1. Hereditary diffuse gastric cancer in a young male with a strong family history of diffuse gastric cancer. A: At a low-power cursory glance the mucosa appears to be more or less normal. B: Higher magnification discloses multiple foci of intramucosal diffuse (signet ring cell) gastric carcinoma. C: E-cadherin immunostain showing staining of the nonneoplastic gastric glands and absence of staining in the tumor cells in the lamina propria and a few isolated tumor cells in the glands (lower right). D: Ki-6 immunostain showing the low proliferative index of the tumor cells. A portion of a nonneoplastic gland shows some proliferation (lower right). |
Interaction between the Environment and Inherited Polymorphisms
By 1966 a large number of studies had found a consistent relationship between the diffuse type of stomach cancer and blood type A (28). It seems unlikely that blood type A has a direct role in the carcinogenic process, but it may serve as a marker for an as yet unidentified mutation, or a genetic polymorphism that increases the cancer risk by modulating the host reaction to environmental hazards. For example, the interleukin-1 (IL-1) B gene that encodes a proinflammatory cytokine is highly polymorphic (29). Gastric cancer patients are more likely to have the IL-1B-31T/IL-1RN* phenotype. Such persons mount an especially strong inflammatory response to H. pylori infection through IL-1 overexpression. The level of gastric cancer risk varies greatly depending on the association of specific genotypes of the infecting organism with different IL-B phenotypes, with an odds ratio (OR) of 87 (CI = 11 to 697) when the vacAs1 strain associates with IL-B-511*T (30). In contrast, when H. pylori strain vacAm1 associates with this phenotype, the OR is substantial but falls to 7.4 (CI = 3.2 to 17). Tumor necrosis factor-α (TNF-α), another proinflammatory protein, is increased in the gastric mucosa of persons infected with H. pylori. The TNF-α-308* allele increases TNF-α production. The risk of acquiring multifocal atrophic gastritis and gastric cancer increases in persons with the high-risk IL-1B and TNF-α genotypes (31).
Variations in glutathione-S-transferase (GST-1), an enzyme that catalyzes the conjugation of numerous carcinogens, may modify the stomach cancer risk from smoking or ingested carcinogens. The null variant of the μ form of GST-1 constitutes 40% of the Japanese population and associates with a modest increased risk of gastric carcinoma (32). A
similar association has been identified between gastric cancer and a variation in the N-acetyl transferase gene (NAT-1) (33). Aromatic and heterocyclic amine carcinogens are acetylated by NAT-1, but persons with a variant form of this enzyme lack this function. Cytochrome 450 2E1 (CYP2E1) is involved in the metabolism of environmental carcinogens. Two reports suggest that polymorphisms in this gene interact with smoking to increase the risk of gastric cancer (34). Finally, the lining of the stomach is protected from environmental insults by mucin. The MUC1 gene is polymorphic and individuals with small MUC1 genotypes have an increased risk of gastric cancer (35).
similar association has been identified between gastric cancer and a variation in the N-acetyl transferase gene (NAT-1) (33). Aromatic and heterocyclic amine carcinogens are acetylated by NAT-1, but persons with a variant form of this enzyme lack this function. Cytochrome 450 2E1 (CYP2E1) is involved in the metabolism of environmental carcinogens. Two reports suggest that polymorphisms in this gene interact with smoking to increase the risk of gastric cancer (34). Finally, the lining of the stomach is protected from environmental insults by mucin. The MUC1 gene is polymorphic and individuals with small MUC1 genotypes have an increased risk of gastric cancer (35).
A recent Korean study has shown that polymorphisms in the DNA repair gene XRCC1 may either enhance or reduce the risk of gastric cancer in this high-risk population (36). Carriers of one haplotype (194/Trp, 280/Arg, 399Arg) are at decreased risk of gastric cancer, while those with another haplotype (194/Arg, 280/Arg, 399Arg) are at increased risk of antral cancer. The polymorphism in XRCC1 interacts with a polymorphism in adenosine diphosphate ribosyl transferase (ADPRT) in such a way that ADPRT and XRCC1 polymorphisms confer host susceptibility to gastric carcinoma possibly from reduced ADPRT–XRCC1 interactions and attenuated base excision repair, particularly in smokers (36a).
Other Genetic Factors
In addition to the genetic factors listed above, there are a number of somatic alterations that play a role in gastric cancer development. These include sporadic alterations in oncogenes, tumor suppressor genes, and mismatch repair genes. These genetic alterations include many that are altered in other cancers. Genetic instability, CpG island methylation, telomerase activation, and p53 mutations tend to occur early in the development of gastric cancer. Some of the more common alterations are listed in Table 5.5.
Epigenetic alterations, the most common of which is CpG island methylation of the promoter regions of cancer-related genes, are also common in gastric carcinoma. There is global DNA methylation and subsequent gene silencing in gastric cancers and its precursors and tumors showing these changes fall into the CpG island methylator (CIMP) phenotype. When the CIMP phenotype occurs, there is frequently an absence of mutation in genes that are commonly mutated in gastric cancer, such as p53 (36b). Genes that are frequently hypermethylated in the setting of Ebstein-Barr virus (EBV) include p16 and hMLH1 (36c). Promoter methylation of E-cadherin is common in the setting of H. pylori infection (36d). Sonic hedgehog, a gene important in foregut development, is also frequently hypermethylated (36e).
Environmental Factors
Environmental factors may affect gastric cancer risk directly or inversely. Several environmental hazards combine to induce distal stomach cancer; other environmental factors protect the stomach from exposure to these hazards. The risk of acquiring stomach cancer ultimately depends on which prevails.
TABLE 5.5 Selected Genetic Alterations in Sporadic Gastric Carcinoma | ||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||
Helicobacter pylori Infection
H. pylori infection is usually the step leading to the development of gastric cancer. The infection is acquired in childhood (37). Infected children carry the infection and its anatomic consequences into middle and old age (9). H. pylori gastritis may affect 75% of persons in high-risk populations, but no more than 5% of infected individuals develop stomach
cancer (38), indicating that infection does not act alone. Prospective studies have shown a consistent association between the presence of distal gastric cancer and high antibody levels against H. pylori (39). Asymptomatic persons with the highest H. pylori antibody levels are the most likely to develop gastric cancer in these studies. When compared to other H. pylori strains, the cagA strain is associated with heightened antibody levels, a more robust inflammatory response, and a higher risk of gastric cancer (39). One mechanism by which H. pylori infection may increase stomach cancer risk is through exposure of replicating cells to reactive oxygen species derived from the inflammatory cells that are part of H. pylori gastritis (Fig. 5.2).
cancer (38), indicating that infection does not act alone. Prospective studies have shown a consistent association between the presence of distal gastric cancer and high antibody levels against H. pylori (39). Asymptomatic persons with the highest H. pylori antibody levels are the most likely to develop gastric cancer in these studies. When compared to other H. pylori strains, the cagA strain is associated with heightened antibody levels, a more robust inflammatory response, and a higher risk of gastric cancer (39). One mechanism by which H. pylori infection may increase stomach cancer risk is through exposure of replicating cells to reactive oxygen species derived from the inflammatory cells that are part of H. pylori gastritis (Fig. 5.2).
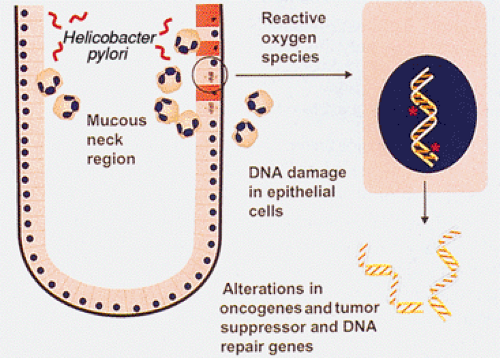 FIG. 5.2. Mechanisms by which Helicobacter pylori increases gastric cancer risk. |
The initial manifestation of H. pylori infection is severe superficial gastritis, followed by the development of multifocal atrophic gastritis and intestinal metaplasia (see Chapter 4). The process begins at the antral–corpus junction and along the lesser curvature of the antrum. Over time the atrophic foci expand and fuse so that in old age much of the oxyntic mucosa is replaced by intestinalized tissue. Cancer risk is highest in H. pylori–infected persons at the nadir of acid production (40). The achlorhydria favors a bacterial flora that may generate carcinogens through the nitrosation of dietary amines (41).
Diet
The pattern of food consumption may increase or reduce the risk of acquiring stomach cancer. The addition of nitroso compounds to the diet of experimental animals produces gastric cancer and its precursor, intestinal metaplasia (42). Epidemiologic studies show a direct association between the development of intestinal metaplasia and nitrate/nitrite consumption in humans (43). Salt intake also directly relates to stomach cancer risk in humans (44). The consumption of dried, salted fish in Japan and the consumption of salted, nitrosated foods through the winter months in northern countries are examples of dietary practices that increase the stomach cancer risk. In contrast, fresh fruits and vegetables provide antinitrosating effects that protect the stomach from both exogenous and endogenous mutagenic nitroso compounds. Fresh fruits and vegetables function as antioxidants and contain substantial amounts of folate, ascorbic acid, carotene, and tocopherol. Epidemiologic studies of Japanese and European populations suggest that raw green and yellow vegetables protect against the development of gastric cancer (45), and a Japanese cohort study showed that increased fresh produce consumption and reduced consumption of pickled food reduced the gastric cancer risk, even in the presence of atrophic gastritis (46). The year-round availability of fresh produce eliminates the need to use salt to preserve vegetables. Smoking and salting of meat is no longer a necessity with the advent of universal household refrigeration. These two factors have contributed to the dramatic decrease in stomach cancer rates in Western countries after World War II.
Smoking
A meta-analysis of 37 case control studies on the relationship of smoking to gastric cancer yielded inconsistent results (47). This inconsistency is surprising and contrasts with the strong association of smoking with gastric ulcer (48), a disease that is closely related to intestinal metaplasia, H. pylori gastritis, and gastric cancer. The same analysis, however, found that each of ten prospective cohort studies showed a significantly increased risk of gastric cancer in the order of 1.5 to 2.5, four of the studies showing a dose response. Thus, the magnitude of smoking-related risk is low, but may account for 11% of stomach cancers worldwide (47).
Radiation
A dose-dependent increase in gastric cancer frequency has occurred among Japanese atomic bomb survivors, suggesting that ionizing radiation may play a role in gastric carcinogenesis (49). Additionally, radiotherapy in young patients increases their risk of gastric cancer when lymph nodes of the upper abdomen are included in the radiation field, as may occur in the treatment of lymphoma and testicular cancer (50).
Previous Gastric Surgery
The creation of a gastroenterostomy after subtotal gastrectomy for peptic ulcer is a well-established risk factor for the development of cancer in the gastric stump (Fig. 5.3) (51). The cancer may not be apparent until 17 to 20 years after the creation of the anastomosis. It is preceded by the appearance of gastritis and hyperplastic polyps at the anastomotic line (52). The cause of the increased risk is attributed to the reflux of bile and pancreatic juice into the gastric remnant, a hypothesis that is supported by rodent experiments that show the sequential development of gastritis, hyperplasia, metaplasia, and adenocarcinoma after duodenal contents
were diverted into the stomach (53). Since subtotal gastrectomy is no longer commonly employed to treat peptic ulcer, this risk factor will disappear in the near future. Its place may be taken by subtotal gastrectomy for early antral cancer, a procedure that has become quite common in Asian high-risk countries with active screening programs of asymptomatic subjects (54). The interval between surgery for early cancer and the development of stump cancer may be shorter than after peptic ulcer surgery. Another potential source of postenterostomy stomach cancer is the Roux-en-Y gastrojejunostomy used to treat morbid obesity. Gastric cancer has been reported in four women at 5, 5, 13, and 22 years after surgery (55). Since bariatric surgery has become a very common procedure, we may expect increasing numbers of such cancers. The defunctionalized gastric segment is subject to the reflux of bile and alkaline secretions and the subsequent development of gastritis (56).
were diverted into the stomach (53). Since subtotal gastrectomy is no longer commonly employed to treat peptic ulcer, this risk factor will disappear in the near future. Its place may be taken by subtotal gastrectomy for early antral cancer, a procedure that has become quite common in Asian high-risk countries with active screening programs of asymptomatic subjects (54). The interval between surgery for early cancer and the development of stump cancer may be shorter than after peptic ulcer surgery. Another potential source of postenterostomy stomach cancer is the Roux-en-Y gastrojejunostomy used to treat morbid obesity. Gastric cancer has been reported in four women at 5, 5, 13, and 22 years after surgery (55). Since bariatric surgery has become a very common procedure, we may expect increasing numbers of such cancers. The defunctionalized gastric segment is subject to the reflux of bile and alkaline secretions and the subsequent development of gastritis (56).
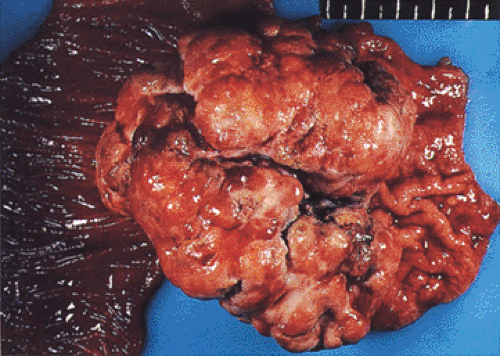 FIG. 5.3. Large polypoid gastric cancer originating in the gastric stump. The small bowel mucosa is seen on the left. |
Predisposing Gastric Lesions
Gastric cancer does not arise de novo from a normal mucosa. Gastritis is usually the first step in cancer induction, whatever the underlying cause. In persons with polymorphisms that render them especially vulnerable to exogenous or endogenous carcinogens, an intense superficial gastritis may be the only anatomic precursor to cancer induction.
Multifocal Atrophic Gastritis and Intestinal Metaplasia
The intestinal-type cancer so characteristic of high-risk populations is preceded by a sequence of changes that begins with inflammation and passes through atrophy and intestinal metaplasia (IM) to dysplasia and ultimately to invasive cancer (57) (Figs. 5.4 and 5.5). These changes are initiated in childhood by H. pylori infection. The dense leukocytic response that affects the superficial lamina propria and the epithelium of the mucous neck region of the gastric glands, as discussed in Chapter 4, is followed by the appearance of foci of atrophic, intestinalized mucosa at the antral–corpus junction. These changes become apparent in adolescents and young adults. The metaplastic glands and the mucosa adjacent to them show increased cell proliferation. The foci of multifocal atrophic gastritis (MAG) enlarge, fuse, and expand proximally and distally so that, by the 6th or 7th decade of life, all but a small portion of the proximal greater curvature of the corpus may be lined by intestinalized mucosa. Two forms of IM are recognized: The “complete” form faithfully reproduces small intestinal-type cells. There are subsite-dependent variations that diverge from this pattern. Some intestinalized glands lack Paneth cells, lose the ability to make some glycocalyceal enzymes, or may produce mucins similar to those of the colon, the so-called “incomplete” form of IM (58). There is a widely held view that incomplete IM carries a higher risk of carcinoma than the complete form, but the most common site for early cancer is the antral–corpus junction, where the complete form predominates. The incomplete form is best seen on the distal greater curvature of the antrum, an area that is involved in the later stages of intestinalization. Cancer risk increases with the extent of the IM; the presence of incomplete IM is synonymous with extensive IM.
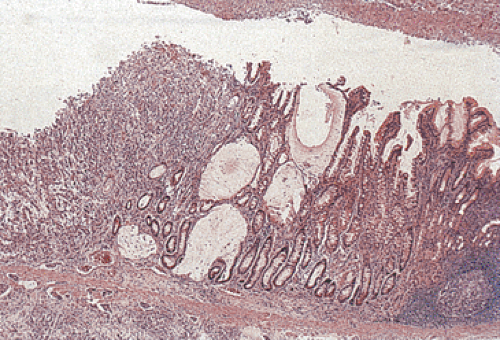 FIG. 5.4. Neoplastic progression in the stomach. A prominent area of follicular gastritis is present on the right side of the photograph. There is a transition to intestinal metaplasia as one moves toward the center of the figure. Dysplasia develops in the areas of intestinal metaplasia, until finally an invasive carcinoma of the intestinal type develops as seen on the far left side of the photograph. Note the tumor within the lymphatics under the muscularis mucosae. |
The mechanisms that underlie the development of IM in MAG are complex. The development and maintenance of specific organ structures and functions in the gastrointestinal tract are regulated by CDX, a homolog of the Drosophila caudal gene, during intestinal development (59). CDX1 and CDX2 are not expressed in the normal stomach, but their mRNAs are expressed in metaplastic glands. CDX2 precedes CDX1 expression and appears to trigger IM. Transgenic mice that express CDX2 in gastric parietal cells have complete replacement of gastric mucosal glands by the full array of intestinal cells,
including goblet cells and absorptive cells (60). A mechanism for the activation of this gene has been suggested by Houghton and Wang (61). They found that, in the presence of H. pylori– generated gastritis, circulating bone marrow stem cells are recruited and engrafted into the replicating zone of the gastric mucosa. The presence of these stem cells in the gastric mucosa in patients with H. pylori gastritis may account for the extreme heterogeneity of gastric cancer and the presence of cells with a hybrid intestinal/gastric phenotype (62,63).
including goblet cells and absorptive cells (60). A mechanism for the activation of this gene has been suggested by Houghton and Wang (61). They found that, in the presence of H. pylori– generated gastritis, circulating bone marrow stem cells are recruited and engrafted into the replicating zone of the gastric mucosa. The presence of these stem cells in the gastric mucosa in patients with H. pylori gastritis may account for the extreme heterogeneity of gastric cancer and the presence of cells with a hybrid intestinal/gastric phenotype (62,63).
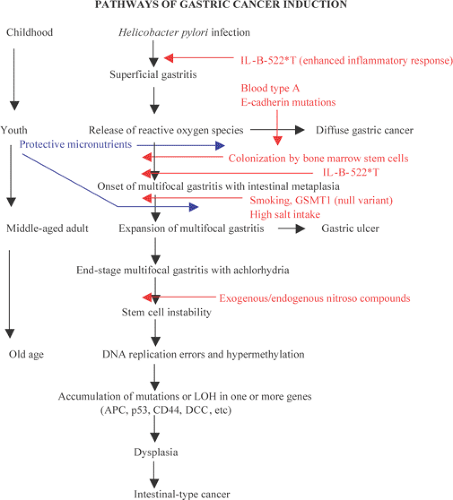 FIG. 5.5. Diagram of the pathways of gastric cancer induction. LOH, loss of heterozygosity. |
Serum pepsinogen levels reflect the extent of gastric mucosal intestinalization. Pepsinogen is a proenzyme activated by gastric acid to produce pepsin. It is produced in two forms: Pepsinogen group I (PGI) is only produced in the oxyntic mucosa, while pepsinogen group II (PGII) is made throughout the stomach and in Brunner glands. Replacement of oxyntic mucosa by IM results in decreasing PGI serum levels (64). A PGI level <30 ηg/mL or a PGI:PGII ratio <2 are established cut-points used in screening programs to identify patients with extensive IM and who are at especially high risk of gastric cancer (65). The predictive value of the PGI level is improved when it is combined with the H. pylori serum antibody levels, as shown in Table 5.6, adapted from a case control study (66). Similar trends are observed with the PGI:PGII ratio.
Autoimmune Gastritis
Autoimmune gastritis, as discussed in Chapter 4, is a well-recognized, but less common, gastric cancer precursor than MAG. Autoimmune gastritis and H. pylori gastritis may coexist in most patients with pernicious anemia and the titers of both antibodies inversely relate to disease duration (67). It may be distinguished from late-stage MAG by the absence of antral inflammation and atrophy. The cancers associated with this form of gastritis arise in the antrum or the atrophic corpus, with a relative risk ranging from 2.2 to 5.6 (68). The increased risk stems from several factors: (a) Loss of gastric acidity favors the growth of bacteria that may generate endogenous nitroso compounds from dietary amines. (b) Gastrin production is greatly increased in response to prolonged achlorhydria. The trophic effects of hypergastrinemia result in accelerated cell turnover in a replicating compartment already expanded due to the loss of parietal and chief cells. (c) These replicating cells are exposed to reactive oxygen species from inflammatory cells.
Gastric Ulcer
A stage is reached in the early phases of the expansion of MAG when the proximal antral intestinalized mucosa is
exposed to the acid produced by the still intact oxyntic mucosa. The intestinalized mucosa lacks the protective mucous barrier of the normal antrum, so that it is vulnerable to peptic ulceration. Patients with gastric ulcer are at greatly increased risk of gastric cancer. This is not surprising since gastric ulcer and gastric cancer share the same risk factors: H. pylori gastritis, a diet rich in salt, smoking (48), and a declining frequency in Western countries (69). As may be expected among patients who have retained the ability to make acid, the mean age of gastric ulcer patients is 10 years younger than the mean age of patients with cancer. Re-epithelialization of the ulcer associates with expansion of the replication zone of the glands bordering the ulcer, exposing a larger number of vulnerable proliferating cells to genotoxic hazards. In contrast, the regenerating epithelial cells that migrate over the ulcer base are arrested in the postmitotic phase of the cell cycle, accounting for the observation that the gastric mucosa bordering the ulcer, rather than the ulcer base, is a common site of early cancer.
exposed to the acid produced by the still intact oxyntic mucosa. The intestinalized mucosa lacks the protective mucous barrier of the normal antrum, so that it is vulnerable to peptic ulceration. Patients with gastric ulcer are at greatly increased risk of gastric cancer. This is not surprising since gastric ulcer and gastric cancer share the same risk factors: H. pylori gastritis, a diet rich in salt, smoking (48), and a declining frequency in Western countries (69). As may be expected among patients who have retained the ability to make acid, the mean age of gastric ulcer patients is 10 years younger than the mean age of patients with cancer. Re-epithelialization of the ulcer associates with expansion of the replication zone of the glands bordering the ulcer, exposing a larger number of vulnerable proliferating cells to genotoxic hazards. In contrast, the regenerating epithelial cells that migrate over the ulcer base are arrested in the postmitotic phase of the cell cycle, accounting for the observation that the gastric mucosa bordering the ulcer, rather than the ulcer base, is a common site of early cancer.
TABLE 5.6 Age-, Sex-, and Ethnicity-adjusted Odds Ratios for Serum Pepsinogen Group I (PGI) Levels and Helicobacter pylori (HP) Antibody Status, According to Histologic Types of Gastric Cancer | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
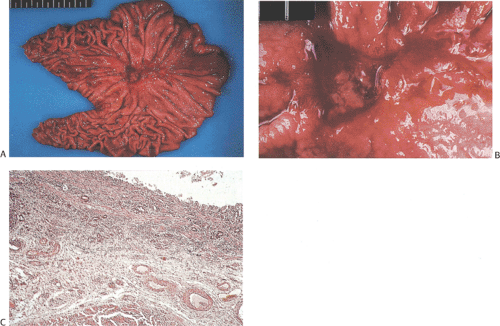 FIG. 5.6. Ulcer cancer. A: Large, centrally located gastric ulcer. B: Closer view. C: Ulcer base. Malignant glands are within and below the muscularis mucosae. |
The term ulcer cancer defines a gastric carcinoma that arises in a pre-existing peptic ulcer (Figs. 5.6 and 5.7). Tumors arising in this setting account for <1% of all gastric
carcinomas. In order to be accepted as an ulcer cancer, the case must show definite evidence of pre-existing chronic peptic ulcer and evidence of coexisting malignancy. About 5% of endoscopically benign ulcers eventually prove to be malignant, but some lesions may require more than one biopsy to detect the underlying malignancy (70). Underestimation of the depth of invasion may occur when the tumor develops from the epithelium of a healed ulcer in which the submucosa and muscularis propria are no longer recognizable because of scarring.
carcinomas. In order to be accepted as an ulcer cancer, the case must show definite evidence of pre-existing chronic peptic ulcer and evidence of coexisting malignancy. About 5% of endoscopically benign ulcers eventually prove to be malignant, but some lesions may require more than one biopsy to detect the underlying malignancy (70). Underestimation of the depth of invasion may occur when the tumor develops from the epithelium of a healed ulcer in which the submucosa and muscularis propria are no longer recognizable because of scarring.
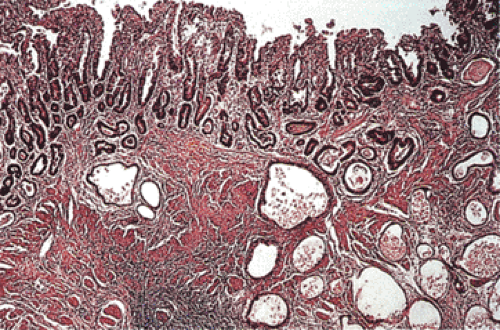 FIG. 5.7. Invasive carcinoma in healed gastric peptic ulcer. Well-differentiated adenocarcinoma infiltrates the muscularis propria, which is fused with the muscularis mucosae. |
Hyperplastic Polyps
Hyperplastic polyps used to constitute from 75% to 90% of all gastric polyps (71). They arise from the foveolar hyperplasia that frequently accompanies atrophic gastritis, whether of the multifocal or autoimmune type. They may also appear on the gastric side of gastroenterostomy stomas. Patients with hyperplastic polyps are at increased risk of gastric cancer, but the cancers very rarely arise in them. Rather, they are byproducts of the chronic gastritis that is the underlying cause of cancer induction. In the unusual event that a cancer does arise in a hyperplastic polyp, it is preceded by the development of dysplasia (Fig. 5.8).
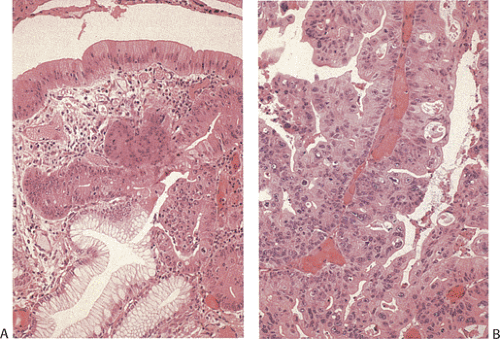 FIG. 5.8. Dysplasia arising in a hyperplastic polyp. A: Low-grade dysplasia spreads from the surface to the deeper portions of gland, replacing the normal foveolar epithelium. B: Higher power view of another area of high-grade dysplasia. The cells are hyperchromatic, pleomorphic, and disorganized. The glands are beginning to develop a back-to-back arrangement suggestive of intramucosal carcinoma. |
Dysplasia
Dysplasia is the first microscopically detectable anatomic change in the neoplastic process. The dysplastic changes may represent cytologic alterations or disorganized architecture. Gastric dysplasia shows two major growth patterns: A flat, grossly inconspicuous dysplasia or polypoid lesions recognizable as adenomas. The dysplastic cells may be gastric or intestinal in type, or they may be hybrid forms from each lineage.
Nonpolypoid Dysplasia
Areas of mucosal dysplasia are characterized by histologic and cytologic abnormalities that must be distinguished from regenerative (reactive) atypia. Atypical regenerative changes are usually accompanied by active inflammation without significant architectural or differentiation abnormalities (Fig. 5.9). In contrast, dysplastic cells show one or more nuclear abnormalities (increased size, hyperchromasia, irregular shape, abnormal mitoses) and form abnormal glands with branching and occasional back-to-back configuration (Figs. 5.10 and 5.11) (71). The identification of dysplasia is critical to the management of patients in high-risk populations, but the assignment of grades is subjective, with wide inter- and intravariations in grade selection. A workshop convened to resolve international differences in the
definitions of different grades of gastric epithelial dysplasia resulted in the Padova classification (72), as summarized in Table 5.7.
definitions of different grades of gastric epithelial dysplasia resulted in the Padova classification (72), as summarized in Table 5.7.
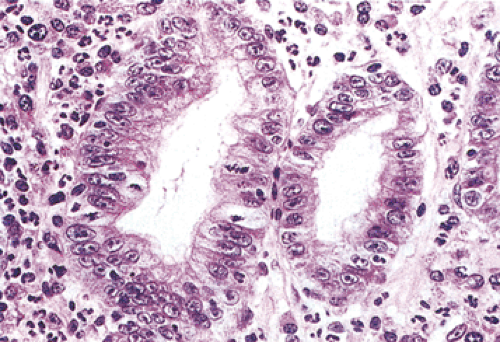 FIG. 5.9. Regenerative hyperplasia. An increased nuclear-to-cytoplasmic ratio and mitotic rate, along with large nucleoli and stromal inflammatory cells, are all present. |
TABLE 5.7 The Padova Classification of Gastric Dysplasia and Related Lesions | |
|---|---|
|
Pathologists achieve fairly high levels of agreement in the diagnosis of high-grade dysplasia, but the agreement is only “fair” for low-grade dysplasia and “poor” for indefinite for dysplasia (73). We believe that it is sufficient to identify dysplasia and to classify it as high or low grade. In low-grade dysplasia, the gastric mucosal architecture is generally preserved, although abnormalities sometimes occur, including pseudovilli, irregular branching papillary infoldings, crypt lengthening with serration, and cystic changes (Fig. 5.10). Dystrophic goblet cells may also be seen. The pyloric glands branch at the crypt base and range from normal in appearance to frankly dysplastic. We have also seen cases in which dysplasia occurred in gastric foveolar cells (Fig. 5.12). The term high-grade dysplasia replaces “carcinoma in situ.” Severe gastric dysplasia shows nuclear stratification, increased abnormal mitoses, disordered polarity, and glandular crowding. Problems associated with diagnosing gastric epithelial dysplasia are threefold: (a) one must be able to separate dysplasia from atypical regenerative changes, (b) one should be able to characterize high-grade from low-grade dysplasia, and (c) the dysplasia should be separable from invasive carcinoma. Immunohistochemical detection of p53 overexpression and Ki-67 (Fig. 5.13) stains to detect expansion of cell proliferation to the mucosal surface and abnormal tumor suppressor gene function may help to distinguish dysplastic from atypical regenerative changes.
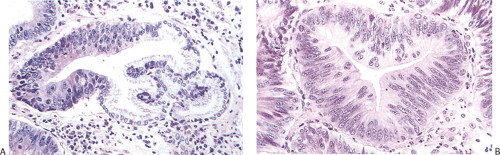 FIG. 5.10. Gastric dysplasia. A: The gastric gland is partially replaced by neoplastic epithelium. B: Mild dysplasia as illustrated here is characterized by cellular crowding and elongation, generally basally located nuclei, few mitoses, little cellular stratification, and nuclear pleomorphism. |
Gastric Adenomas
Gastric adenomas are less common than hyperplastic polyps and, unlike colonic adenomas, they are uncommon in the
absence of FAP. They may be flat (Fig. 5.14) or pedunculated (Fig. 5.15). An endoscopic study in high-risk Korean patients found that 74.5% of these lesions arose in the distal third of the stomach. Focal areas of malignancy were found in 6.7% of adenomas, all but one arising in the distal stomach (74).
absence of FAP. They may be flat (Fig. 5.14) or pedunculated (Fig. 5.15). An endoscopic study in high-risk Korean patients found that 74.5% of these lesions arose in the distal third of the stomach. Focal areas of malignancy were found in 6.7% of adenomas, all but one arising in the distal stomach (74).
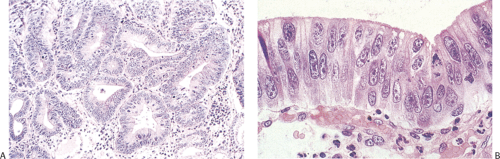 FIG. 5.11. Severe dysplasia. A: Gastric dysplasia with distorted architecture and severely dysplastic epithelium. There are numerous mitoses. B: The cells are so stratified that the nuclei reach the luminal surface. |
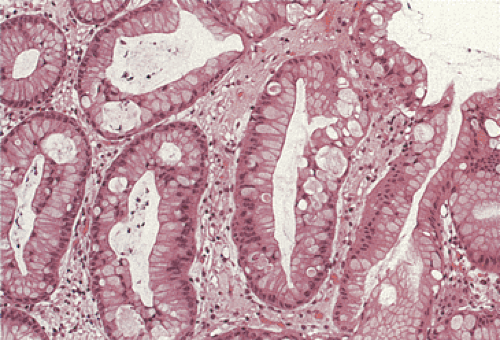 FIG. 5.12. Foveolar dysplasia. The foveolar epithelium is disorganized with some loss of polarity and the nuclei are pseudostratified. No inflammation is present. |
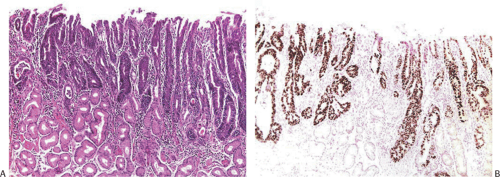 FIG. 5.13. High-grade dysplasia replacing the gastric pits. A: Hematoxylin and eosin–stained section. B: p53 immunostain shows nuclear labeling in virtually all of the neoplastic cells. |
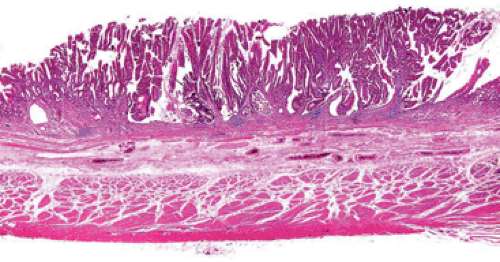 FIG. 5.14. Flat villous adenoma. |
Gastric and colon polyps share some features. Grossly they have a lobulated or mamillated surface and are often covered by a reddened, velvety mucosa (Fig. 5.15). Abnormal epithelium occupies the surface and the luminal portions of the pit. With increasing degrees of dysplasia there is cellular stratification, progressive loss of nuclear polarity, and increased cellular crowding. The nuclei appear close to the lumina of the tubules and eventually secondary lumina form. A villous configuration may predominate in larger lesions (Fig. 5.14). When traumatized, adenomas may acquire surface changes resembling those seen in hyperplastic polyps. Gastric adenomas contain many dysplastic cell types, including enterocytes, goblet cells, endocrine cells, and Paneth cells. The World Health Organization (WHO) classification subdivides gastric adenomas into three subtypes: Tubular, papillary, and papillary–tubular. In contrast, Japanese authors recognize two types of adenoma: Protruding and depressed (75). Depressed adenomas (Fig. 5.16) are significantly larger than protruding adenomas and are more likely to contain high-grade dysplasia than protruded adenomas. We have also seen serrated adenomas arising in the stomach (Fig. 5.17). These lesions are far too rare to know whether this morphology reflects the underlying genetic alterations as it does in the colon (see Chapter 14).
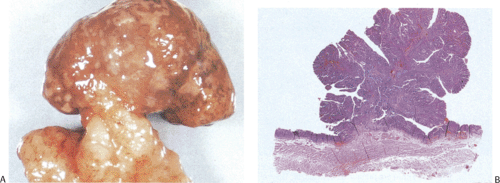 FIG. 5.15. Gastric adenoma. A: The pedunculated adenoma is covered by a reddened mucosa. B: Whole mount section of the adenoma depicted in A showing the proliferation of the neoplastic cells. |
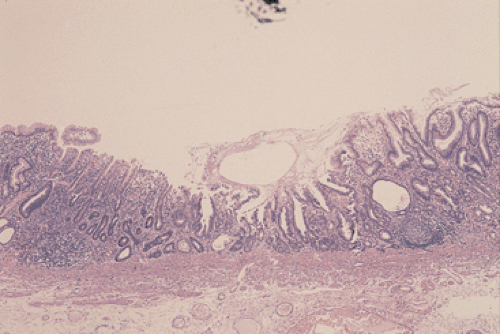 FIG. 5.16. Depressed adenoma. The adenoma appears as the central mucosal depression. |
Histogenesis of Gastric Cancer
Areas of dysplasia may remain stable, regress (76), or take several years to progress to invasive intestinal-type cancer (77). In contrast, invasive diffuse cancers in young persons may arise in a mucosa that does not appear overtly dysplastic (Fig. 5.1), although in situ carcinoma may be present as discussed below. The factors that determine the cancer phenotype and the speed of its progression from normal gastric epithelium are complex. An invasive cancer might arise directly from the replicating compartment in the neck region of gastric glands, from the crypts of intestinalized glands, or from replicating surface cells in areas of dysplasia. The cells
of origin may determine the phenotype of an early cancer, but postinduction mutations may introduce so much heterogeneity that the mother cells of advanced cancers cannot be determined. The types and numbers of genetic and epigenetic changes in the cancer influence the speed of its progression from an early superficial lesion to a widely disseminated one.
of origin may determine the phenotype of an early cancer, but postinduction mutations may introduce so much heterogeneity that the mother cells of advanced cancers cannot be determined. The types and numbers of genetic and epigenetic changes in the cancer influence the speed of its progression from an early superficial lesion to a widely disseminated one.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


