
The Kidney in Hypertension
Global Burden of Hypertension
Cardiovascular disease is the most common cause of death in economically developed countries and is rapidly evolving as a major cause of morbidity and mortality in economically developing nations as well (1). Certainly, hypertension is among the most important modifiable risk factors and the leading risk factor for disease burden worldwide (2). On the basis of currently recommended criteria for diagnosis of hypertension (systolic blood pressure [SBP] ≥140 mm Hg or diastolic blood pressure [DBP] ≥90 mm Hg, or use of antihypertensive medication), approximately one-third of men and women have hypertension in the United States (3). The overall prevalence of hypertension has not changed since 1999 to 2000 (4). Recent changes to Joint National Committee (JNC) 8 proposed less restricted guidelines for BP targets in individuals with hypertension (5). Despite this, BP goals were met in less than half of the young adults and two-thirds of older adults in the United States (6). There is a progressive rise in SBP throughout life, with a difference of 20 to 30 mm Hg between early and late adulthood. Likewise, DBP tends to rise with age up until the fifth decade; in later decades DBP declines. These patterns result in a progressively higher prevalence of hypertension with aging, which consists of predominant elevation of SBP or isolated systolic hypertension. A majority of adults have hypertension by the sixth decade and >70% have it by the seventh and eight decades of life. Among normotensive individuals in their sixth decade, the lifetime risk of developing hypertension approaches 90%. African American adults have an incidence and prevalence of hypertension that is 50% higher than their white or Mexican American counterparts. The average level of BP and prevalence of hypertension also increased progressively in children and adolescents between 1988 and 2000. The prevalence of hypertension in many other countries is as high as or higher than that identified in the United States. Estimates by the World Health Organization (WHO) suggest that approximately 40% of the adults have hypertension, with the highest prevalence in Africa estimated at approximately 46% (7). In the last few decades, the prevalence of hypertension in economically developing countries appears to have risen; in 2008, 1 billion individuals had uncontrolled hypertension (7). BP exhibits a dose-dependent relationship with the risk of cardiovascular disease throughout the entire range of BP in the population. The relationship is independent of other cardiovascular risk factors with no evidence of a BP threshold for risk. The BP risk applies to all major manifestations of cardiovascular disease including stroke, sudden cardiac death, coronary heart disease, heart failure, aortic aneurysm, peripheral vascular disease, as well as chronic kidney disease (CKD) and end-stage renal disease (ESRD). Although elevations of both SBP and DBP are independently associated with cardiovascular risk, high SBP is the most potent predictor of risk (8,9). Population-attributable risk estimates had indicated that attainment of a population average SBP <115 mm Hg would reduce the occurrence of ischemic heart disease by 50% and stroke by 60%, thereby preventing about 7 million deaths annually on a worldwide basis (1).
Historical Perspective: The Link between Hypertension and Renal Dysfunction
The occurrence of hypertension in the setting of renal disease and its impact on the progression of renal insufficiency has long been of interest to clinicians. The concept that hypertension is in some way related to renal dysfunction was first proposed by Bright in 1836 (10). He recognized the association between hypertrophy of the heart and contraction of the kidney and postulated that the cause was increased cardiac work required to force blood through a vascular tree constricted by irritating humoral substances that accumulate in renal failure. The role of fluid retention in the genesis of renal hypertension was first outlined by Traube in 1871 (11), who proposed that with shrinkage of the renal parenchyma, a decreasing amount of fluid is removed from the arterial system by urinary secretion, thereby resulting in hypertension.
Mahomed in 1879 (12) was the first to clearly describe hypertension of unknown cause, without evidence of underlying renal disease (now called essential hypertension). He emphasized that the most frequent complications in individuals with this type of hypertension were cardiovascular and most often occurred in the absence of significant renal dysfunction. However, in 1914, Volhard and Fahr (13) defined a subgroup of patients with essential hypertension who eventually developed severe renal involvement. They distinguished two types of hypertensive nephrosclerosis, benign and malignant. The benign type, characterized by hyaline arteriolosclerosis, was associated with a slowly progressive course with eventual complications caused by heart failure or stroke in the absence of clinically significant renal impairment. In contrast, malignant nephrosclerosis was characterized by arteriolar necrosis and endarteritis that resulted in rapidly progressive renal failure and death. Volhard (14) subsequently introduced the concept of the vicious circle in which renal disease causes hypertension, which in turn exacerbates renal injury.
Over the years, it has been recognized that the kidney is both “villain and victim” in hypertension (15). The kidney, even when histologically normal, is felt to play a central role in the pathogenesis of essential hypertension. Molecular genetic studies have identified mutations in eight genes that cause mendelian forms of hypertension (16). Each of these genetic defects that impart very large effects on BP leads to enhanced renal tubular sodium reabsorption (impaired natriuresis), resulting in salt-sensitive hypertension. Recently, two coding sequence variants in apolipoprotein L1 (ApoL1), G1 and G2, have been identified as an important risk factor for progressive nondiabetic kidney disease associated with hypertension in African Americans (17). In addition, other underlying primary renal parenchymal disease or abnormalities of the renal vasculature can cause secondary hypertension. On the other hand, the kidney may also suffer the brunt of hypertension. Essential hypertension that enters a malignant phase can rapidly destroy the kidney. Furthermore, recent evidence suggests that hypertension is a major factor in the progression of CKD in the setting of both diabetic and nondiabetic CKD.
We address three major questions with regard to the interaction between the kidney and hypertension. What role does the kidney play in the genesis of essential hypertension and the various forms of secondary hypertension? Why does hypertension develop in the setting of primary renal disease? What is the role of treatment of hypertension in slowing the progression of CKD?
Circulatory Hemodynamics: Complexity in a Simple Relationship
At face value, the physiology of BP regulation is deceptively simple, behaving according to Ohm’s law whereby the mean arterial pressure (MAP) is defined by the product of the cardiac output times the systemic vascular resistance. Thus, hypertension can only result from an increase in either of these two variables. As such, the kidney is expected to have a major influence on BP given its central role in the regulation of extracellular fluid (ECF) volume. An increase in ECF volume caused by renal sodium and water retention should cause increased blood volume, venous return, and filling pressure, which in turn should increase stroke volume and cardiac output via the Frank–Starling mechanism and result in hypertension. Volume expansion, resulting from excess sodium intake or an underlying abnormality in renal sodium excretory mechanisms, has been considered to be an important mechanism for the development of hypertension in animal models as well as in humans with essential hypertension or CKD.
The paradox, which we attempt to explain, is that a significant increase in ECF volume or cardiac output is difficult to demonstrate in human essential hypertension as well as animal models, at least in the chronic phase. Chronic essential hypertension is virtually always maintained by an increase in systemic vascular resistance arising primarily at the level of precapillary arterioles throughout the circulatory system (18). This observation has formed the basis for the widely held concept that an increase in systemic vascular resistance is the underlying primary cause of hypertension, and many theories exist regarding the roles of various vasoconstrictor mechanisms (19) or absence of vasodilator substances (20).
However, there are complexities in the seemingly simple equation that relates BP to cardiac output and systemic vascular resistance. The circulatory system is dynamic and a perturbation, such as ECF volume expansion, which initially leads to hypertension via an increase in cardiac output, may result in compensatory hemodynamic responses that ultimately restore sodium balance and thereby normalize ECF volume and cardiac output. This restoration of sodium balance results from a “pressure natriuresis” caused by systemic hypertension that is maintained by a chronic increase in systemic vascular resistance (21–25). The various theories proposed to account for the phenomenon whereby initially volume-dependent hypertension evolves into high systemic vascular resistance hypertension are described in detail in the following sections.
Dietary Salt and Hypertension
Archeological studies of Paleolithic man suggest that the diet of these hunter-gatherers was very low in sodium and relatively high in potassium. Sodium intake averaged 30 mmol/day with a ratio of dietary potassium to sodium of 16:1 (26). Because total body sodium content is the major determinant of ECF volume, a very efficient renal sodium conservation mechanism evolved in the face of this limited availability of sodium. In contrast, the modern urban diet contains 120 to 300 mmol sodium per day (24) and only 65 mmol potassium per day (27). Therefore, the human species today is faced with a much higher daily sodium load than that to which it adapted over roughly 2 million years (26). Because virtually all ingested sodium is absorbed, sodium intake in excess of insensible losses must be excreted by the kidney. Under normal circumstances, even in the face of tremendous dietary sodium excess, humoral and other mechanisms cause an increase in urinary sodium excretion with little increase in ECF volume (28). However, this natriuretic mechanism is aberrant in a segment of the population, so that excessive dietary salt intake results in the development of hypertension (21–25,29).
EPIDEMIOLOGIC AND CLINICAL STUDIES
Studies throughout the world have suggested a correlation between the mean dietary sodium intake and prevalence of hypertension in the population. In parts of Japan where the mean sodium intake is >400 mmol/day, the prevalence of hypertension approaches 50% (30). In contrast, in certain inland populations where the sodium intake is very low (0.2–51 mmol/day), hypertension is virtually nonexistent and there is no tendency for the BP to rise with age (31). The INTERSALT study investigated the relation between dietary sodium intake (defined by 24-hour urine sodium excretion) and BP in 10,000 subjects in 32 countries (32). A significant positive correlation was found between sodium intake and BP even when the data were adjusted for age, gender, body weight, and alcohol consumption. The data suggest that habitually high sodium intake (>150 mmol sodium per day) is a critical environmental factor that contributes to the high prevalence of hypertension in most urban populations, whereas lifelong ingestion of a diet extremely low in sodium (<50 mmol/day) prevents the development of hypertension (30,31). Extremely high sodium intake (>800 mmol sodium per day) has been shown to raise the BP of healthy normotensive individuals (33). On the other hand, diets with <10 mmol sodium per day have been shown to lower the BP of most hypertensive patients (34). Additional support for the importance of dietary sodium in the genesis of hypertension is the finding that, in an adult population, long-term lowering of the sodium intake was associated with a fall in the prevalence of hypertension and a concomitant reduction in cerebrovascular mortality (35).
The effect of dietary composition on BP is a subject of major public health importance. The Dietary Approaches to Stop Hypertension (DASH) diet study demonstrated that a diet that emphasizes on fruits, vegetables, and low-fat dairy foods; includes whole grains, poultry, fish, and nuts; contains only small amounts of red meat, sweets, and sugar-containing beverages; and has reduced amounts of total and saturated fat and cholesterol substantially lowers BP as compared with the typical diet in the United States (36). In a subsequent report, the effect of different levels of dietary sodium intake, in conjunction with the DASH diet, was studied in subjects with and without hypertension (37). A total of 412 participants were randomly assigned to eat either a control diet typical of intake in the United States or the DASH diet. Within each assigned diet, participants ate foods containing high sodium (150 mmol/day, typical consumption in the United States), intermediate sodium (100 mmol/day, as in the no-added salt diet), and low sodium (50 mmol/day) for 30 consecutive days each, in random order. The DASH diet was associated with significantly lower SBP at each sodium intake level. Moreover, the reduction in sodium intake significantly lowered SBP and DBP in a stepwise fashion, with both the control and DASH diets. Reducing dietary sodium intake had approximately twice as great an effect on BP with the control diet as it did with the DASH diet. Reducing the sodium intake from high to intermediate level reduced the SBP by 2.1 mm Hg (P < 0.001) during the control diet and by 1.3 mm Hg (P = 0.03) during the DASH diet. Reducing sodium intake from intermediate to low level caused additional reductions of 4.6 mm Hg during the control diet (P < 0.001) and 1.7 mm Hg during the DASH diet (P < 0.01). Dietary sodium restriction resulted in a greater reduction in BP in subjects with hypertension than in normotensive subjects. As compared with the control diet with high sodium intake, the DASH diet with low sodium intake led to a mean SBP that was 7.1 mm Hg lower in participants without hypertension and 11.5 mm Hg lower in participants with hypertension. The effects of dietary sodium on BP were observed in participants with and without hypertension, African Americans, and those of other races, and in both men and women. On the basis of the levels of dietary sodium intake actually achieved in the study, it appears that BP can be reduced in individuals consuming either a diet that is typical in the United States or the DASH diet by reducing sodium intake from approximately 140 mmol/day (average sodium intake in the United States) to an intermediate level of 100 mmol/day (the currently recommended no-added salt diet), or from this level to a still lower level of 65 mmol/day (equivalent to 1.5 g sodium or 3.8 g sodium chloride). The impact of these findings on public health of course depends on the ability of people to make long-lasting dietary modifications and increased availability of lower sodium foods in the marketplace.
The Genetic Epidemiology Network of Salt-Sensitivity (GenSalt) study demonstrated that nondiabetic individuals with metabolic syndrome and underlying insulin resistance have increased sensitivity of BP to changes in dietary sodium intake (38). In GenSalt, 1,906 Chinese participants without diabetes, ages 16 years or more, were selected to receive a low-sodium diet (51.3 mmol/day) for 7 days followed by a high-sodium diet (308.8 mmol/day) for an additional 7 days. BP was measured at baseline and on days 2, 5, 6, and 7 of each intervention. Metabolic syndrome was defined as the presence of three or more of the following: abdominal obesity, raised BP, high triglyceride concentration, low high-density lipoprotein (HDL) cholesterol, or high glucose. High salt sensitivity was defined as a decrease in MAP of >5 mm Hg during low-sodium diet or an increase of >5 mm Hg during the high-sodium diet. Overall 283 (28%) of subjects met criteria for diagnosis of metabolic syndrome. Multivariable-adjusted mean changes in BP in response to changes in dietary sodium intake were significantly different in subjects with and without the metabolic syndrome. There was a significant and graded association between the number of risk factors for metabolic syndrome and age-adjusted and gender-adjusted proportion of study participants with high salt sensitivity of BP. Compared with those with no risk factors for the metabolic syndrome, participants with four or five risk factors had a 3.54-fold increased odds of high salt sensitivity during the low-sodium intervention and a 3.13-fold increased odds of high salt sensitivity after the high-sodium diet. Moreover, compared with participants without the metabolic syndrome (less than three risk factors), participants with metabolic syndrome (three or more risk factors) had a 92% increased odds of high salt sensitivity after the low-sodium diet and a 70% increased odds of high salt sensitivity after the high-sodium intervention. The association between metabolic syndrome and salt sensitivity of BP was independent of age, gender, body mass index, physical activity, cigarette smoking, alcohol consumption, and baseline dietary intake of sodium and potassium. Furthermore, the association between metabolic syndrome and salt sensitivity remained significant even if participants with baseline hypertension were excluded. As discussed in more detail later, insulin resistance and concomitant compensatory hyperinsulinemia may lead to renal sodium retention (impaired natriuresis) and thereby explain the BP response to changes in dietary sodium intake in individuals with metabolic syndrome. Data from the GenSalt study suggest that reduction in dietary sodium intake may be an especially important strategy for reducing BP in an individual with multiple risk factors for the metabolic syndrome.
On balance, it is clear that sodium intake must play a permissive role in the development of hypertension, because a lifelong diet very low in sodium prevents hypertension (31). On the other hand, excessive sodium intake alone is not sufficient to cause hypertension, because the majority of individuals on such a diet fail to develop hypertension. These observations imply that there must be additional predisposing factors that lead to the development of hypertension in certain individuals when the intake of sodium is >60 to 70 mmol/day.
DIETARY SALT AND ANIMAL MODELS OF GENETIC HYPERTENSION
When groups of rats were maintained on a wide range of dietary sodium, the mean BP in each group was directly related to sodium intake. Dahl noted that at each level of dietary sodium intake, only some rats became hypertensive. By selective inbreeding, he was able to show that the predisposition to develop hypertension was genetically determined, and he produced a salt-sensitive strain that develops hypertension on a high-sodium diet and a salt-resistant strain that remains normotensive (39).
The Role of the Kidney in Essential Hypertension
In kidney cross-transplantation experiments between four different strains of genetically hypertensive rats (Dahl salt-sensitive hypertensive rats, Milan hypertensive rats, spontaneously hypertensive rats [SHR], and stroke-prone spontaneously hypertensive rats [SHRSP]), and their respective normotensive control strains, it was found that BP determinants were carried within the kidney (40). Thus, transplantation of a kidney from a hypertensive strain rat into a bilaterally nephrectomized rat from the normotensive strain results in hypertension in the recipient. Conversely, transplantation from a normotensive strain rat into a nephrectomized hypertensive strain rat prevents the development of hypertension. Thus, the BP of the recipient rat is dependent on the source of the donated kidney. On the other hand, it could be argued that the posttransplant hypertension in recipients of kidneys from hypertensive strains is not caused by a primary defect in the donor kidney, but instead results from hypertension-induced changes in the donor kidney and that these acquired secondary structural defects in the kidney lead to hypertension in the recipient. To address this question, the development of hypertension in the SHRSP kidney donors was prevented by chronic antihypertensive drug treatment (40). Despite sustained normalization of BP in the donor rats, the recipients developed posttransplant hypertension. This finding indicates that SHRSP kidneys carry a primary defect that can elicit hypertension. These experiments suggest that the predisposition to genetic hypertension resides in the kidney and is not determined directly by systemic humoral abnormalities or changes in vascular reactivity that have been described in these models. The latter abnormalities must represent either epiphenomena or secondary changes in response to a primary renal abnormality.
These animal models of hypertension bear a remarkable resemblance to human essential hypertension. Indeed, studies of kidney transplant patients also support a primary role for the kidney in the development of essential hypertension. In patients with essential hypertension and renal failure caused by malignant nephrosclerosis, bilateral native nephrectomy in conjunction with a well-functioning renal allograft, from a normotensive cadaver donor, cures essential hypertension (41). In a study of six such patients, before renal transplantation, MAP was 168 ± 9 mm Hg despite treatment with a minimum of a four-drug antihypertensive regimen. However, following bilateral native nephrectomy and successful renal transplantation, at a mean follow-up of 4.5 years, MAP without antihypertensive treatment was 92 ± 1.9 mm Hg. Another observation that suggests a role for the kidney in the pathogenesis of hypertension is the finding that the incidence of hypertension in recipients of cadaver kidneys correlates with the incidence of essential hypertension in the family of the donor (42). These intriguing reports support the notion that the defect that causes human essential hypertension resides within the kidney.
Pathogenetic Mechanisms of Impaired Natriuresis
If the relationship between sodium intake and hypertension represents cause and effect, then it is important to explain why high sodium intake leads to hypertension in only some individuals. In this regard, it has been postulated that in the setting of essential hypertension in humans or in salt-sensitive animal models, there is a genetically predetermined impairment in the renal ability to excrete sodium (21–25). This postulated renal abnormality has been termed an “unwillingness to excrete sodium” or “impaired natriuretic capacity.” In studies of isolated, perfused kidneys from Dahl salt-sensitive strains, at age 8 weeks, even before the onset of hypertension, there is a defect in natriuresis such that at any given perfusion pressure, less sodium is excreted in comparison with kidneys from salt-resistant strains (43). In humans, the heritability of essential hypertension has been well established in epidemiologic surveys. The prevalence of hypertension among offspring has been reported to be 46% if both parents are hypertensive, 28% if one parent is hypertensive, and only 3% if neither parent is hypertensive (44). The familial aggregation of hypertension is not simply attributable to shared environmental effects because adoption studies show greater concordance of BP among biological siblings than adoptive siblings living in the same household (45). Moreover, twin studies document greater concordance of BP between monozygotic twins than dizygotic twins (46). Analysis of the natriuretic response to slow infusion of saline has revealed that normotensive first-degree relatives of patients with essential hypertension excrete a sodium load less well than control subjects without a family history of hypertension (47). Among blacks and individuals over 40 years old (48)—two groups with an increased incidence of hypertension—studies of normotensive individuals also have demonstrated a slower natriuretic response to saline infusion than in controls, suggesting that a diminished natriuretic capacity may underlie the predisposition to essential hypertension in these groups.

Figure 9-1 Alterations in renal sodium handling that influence systemic blood pressure (BP). This diagram of the nephron illustrates the molecular pathways mediating sodium reabsorption in individual cells in the proximal tubule (PT) (Na+/H+ exchanger, NHE-3); the thick ascending limb (TAL) of the loop of Henle (Na/K/2Cl cotransporter, NK2CC); the distal convoluted tubule (DCT) (thiazide-sensitive Na/Cl cotransporter, NCC); and cortical collecting duct (amiloride-sensitive epithelial sodium channel, ENaC, the activity of which is regulated by the mineralocorticoid receptor, MR). Also shown is the pathway of the renin–angiotensin–aldosterone system (RAAS), the major regulator of renal salt reabsorption, sodium balance, and BP. Various inherited diseases, medical disorders, and pharmacologic agents that influence renal sodium handling and BP are listed adjacent to the molecular pathways they involve in Figure 9-1. Conditions that enhance renal sodium reabsorption and therefore cause hypertension are labeled HTN. Conditions and drugs that impair renal sodium reabsorption and thereby lower blood pressure are labeled LBP. In the PT, downregulation of NHE-3 mediates acute pressure natriuresis. In the TAL, three different variants of Bartter syndrome result from various autosomal recessive loss-of-function mutations that impair sodium reabsorption and lower blood pressure. Type 1 results from mutation in the Na+/K+/2Cl cotransporter, which is also the transporter inhibited by loop diuretics. Type 2 results from mutation in the ROMK channel, which is crucial for recycling of potassium from the cell into the lumen to maintain efficiency of the NK2CC. Type 3 results from mutation of the basolateral chloride channel, which is responsible for the exit of reabsorbed chloride from the cell. In the DCT, Gitelman syndrome results from autosomal recessive loss-of-function mutation in the NCC, which is also the molecular site of action of thiazide diuretics. Chronic pressure natriuresis is mediated by downregulation of the abundance of the NCC. In the DCT, Liddle syndrome results from gain-of-function mutations in the ENaC leading to enhanced sodium reabsorption and hypertension. ENaC is blocked by the potassium-sparing diuretic amiloride. Loss-of-function mutations in ENaC occur in the autosomal recessive form of pseudohypoaldosteronism type 1 (PHA1). The MR S810L mutation in the MR leads to an autosomal dominant form of hypertension that is markedly exacerbated during pregnancy because the mutant receptor is activated by progesterone. MR is inhibited by spironolactone, another potassium-sparing diuretic. Loss-of-function mutations in the MR cause autosomal dominant PHA 1. In the syndrome of apparent mineralocorticoid excess (AME), loss-of-function mutations in 11-b-hydroxysteroid dehydrogenase (11b-HSD) prevent metabolism of cortisol to cortisone so that cortisol binds and activates the MR leading to enhanced sodium reabsorption and hypertension. Natural licorice causes hypertension by inhibiting 11b-HSD. Cushing syndrome and ectopic secretion of ACTH by certain tumors lead to markedly elevated cortisol levels that overwhelm 11b-HSD, allowing cortisol to bind to MR, leading to enhanced sodium reabsorption and hypertension. Glucocorticoid remediable hyperaldosteronism (GRA) causes hypertension as the result of a chimeric gene that drives constitutive overexpression of aldosterone under the influence of ACTH. Aldosterone synthase deficiency and 21-hydroxylase deficiency (21-OHase def) result in hypotension because of the inability to produce aldosterone and other 21-hydroxylated mineralocorticoids. The adrenogenital syndromes result from deficiencies of 17a-hydroxylase (17a-OHase) or 11b-hydroxylase (11b-OHase), which result in overproduction of the mineralocorticoid hormone deoxycorticosterone (DOC), resulting in enhanced renal sodium reabsorption and hypertension. Increased activity of the renin–angiotensin–aldosterone axis may mediate salt-sensitive hypertension in renovascular hypertension (RVHTN), autosomal dominant polycystic kidney disease (ADPKD), and a subset of patients with essential hypotension who fail to downregulate the RAAS during salt loading (nonmodulators). Additional abbreviations: AI, angiotensin I; ACE, angiotensin-converting enzyme; AII, angiotensin II; HTN, hypertension; LBP, low blood pressure. (Reprinted from Lifton RP, Gharavi AG, Geller DS. Molecular mechanisms of human hypertension. Cell. 2001;104(4):545–556, 2001, with permission from Elsevier.)
NORMAL RENAL SODIUM HANDLING
On a daily basis, the kidneys filter >170 L of plasma containing 23 mol of sodium. Therefore, in an individual consuming a 2-g sodium diet containing 100 mEq of sodium, maintenance of sodium homeostasis requires that the kidneys reabsorb 99.5% of the filtered sodium load. This efficient process of renal sodium reabsorption is accomplished by a complex integrated array of sodium exchangers, sodium transporters, and sodium ion channels. Along the entire length of the nephron, the driving force for sodium reabsorption is the Na+/K+ ATPase located in the basolateral membrane of tubular cells, which extrudes sodium from the cell into the blood-side of the tubule and maintains low intracellular sodium concentration. The distinct functional properties of various portions of the nephron are determined by differences in the sodium transporters located in the apical membrane (Fig. 9-1). Sixty percent of the filtered sodium is reabsorbed in the proximal tubule (PT), largely by the Na+/H+ exchanger (NHE-3) and to a lesser extent by the sodium phosphate cotransporter (NaPi-2). Thirty percent of filtered sodium is reabsorbed in the thick ascending limb (TAL) of Henle by the Na/K/2Cl cotransporter. Seven percent is reclaimed by the thiazide-sensitive Na/Cl cotransporter (NCC) in the distal convoluted tubule (DCT). The remaining 2% to 3% is reabsorbed via the epithelial sodium channel (ENaC) in the cortical collecting tubules. Although ENaC accounts for only a small fraction of total renal tubular sodium reabsorption, this is the principal site for regulation of net sodium balance because the activity of this channel is highly regulated by the renin–angiotensin–aldosterone system (RAAS). Decreased perfusion pressure in the afferent arteriole or decreased delivery of sodium to the TAL leads to secretion of renin, an aspartyl protease that acts on angiotensinogen produced in the liver to produce angiotensin I (AI). Through the action of angiotensin-converting enzyme (ACE) in the lung and elsewhere, AI is converted to angiotensin II (AII). AII binds to its specific G protein-coupled receptor in the zona glomerulosa of the adrenal gland, leading to increased secretion of aldosterone, the principal mineralocorticoid steroid hormone. The actions of aldosterone are mediated chiefly by binding to intracellular nuclear hormone mineralocorticoid receptors (MRs) that function as transcription factors when they are in the activated state (49). Aldosterone stimulates sodium retention by the kidney in part through its action to regulate the ENaC, which mediates apical sodium entry across principal cells in the collecting tubules (50). Aldosterone also stimulates sodium reabsorption in the DCT by increasing the abundance of the thiazide-sensitive NCC (51). Thus, both NCC and ENaC appear to be primary targets for the regulation of sodium excretion by aldosterone.
MOLECULAR MECHANISMS IMPLICATED IN BLOOD PRESSURE VARIATION IN HUMANS
Landmark molecular genetic studies by Lifton et al. recently have identified a substantial number of genes in which rare mutations impart large effects on BP leading to various mendelian forms of hypertension or hypotension (16). Investigation of the genetic causes of hypertension or hypotension has provided major insights into the pathophysiologic mechanisms that can lead to hypertension. Given the vast array of physiologic systems that can influence BP, it is striking that, in all of the mendelian forms of hypertension and hypotension discussed in detail in the following sections, the fundamental underlying abnormality has been found to involve alternations in renal sodium handling (Fig. 9-1). These findings clearly establish the central role of altered sodium homeostasis in the pathogenesis of hypertension and underscore the pivotal role of the kidney in the long-term regulation of BP. Unfortunately, genetic studies in the general population thus far have been disappointing in that no genetic variants that have a substantial effect on BP have been identified. Nonetheless, the finding that all known inherited and acquired forms of hypertension converge on the same final common pathway leading to impaired natriuresis suggests that the pathophysiologic disorders that lead to essential hypertension in the general population will ultimately be found to result directly or indirectly from abnormalities in renal sodium handling.
BLOOD PRESSURE VARIATIONS CAUSED BY MUTATIONS AFFECTING CIRCULATING MINERALOCORTICOID HORMONE
The major regulator of ENaC activity is the MR and its steroid hormone ligand aldosterone. A number of genetic disorders leading to hypertension or hypotension result from abnormalities in aldosterone secretion or the production of other steroid hormones that activate the MR. Glucocorticoid remediable aldosteronism (GRA) is an autosomal dominant trait with the phenotype of early-onset hypertension with normal or elevated plasma aldosterone despite suppressed plasma renin activity (PRA) (Fig. 9-1) (52). Hypokalemia and metabolic alkalosis are present in some patients. The hallmark of this disease is normalization of BP during treatment with exogenous glucocorticoids, which completely suppresses the overproduction of aldosterone. GRA is caused by a gene duplication arising from an unequal crossover between the gene encoding for steroid 11β-hydroxylase (an enzyme involved in cortisol biosynthesis that contains an adrenocorticotropic hormone [ACTH] response element) and the gene encoding for aldosterone synthase (the rate-limiting enzyme in aldosterone biosynthesis in the adrenal glomerulosa). The resulting chimeric gene encodes for a protein with aldosterone synthase enzymatic activity whose expression is regulated by ACTH. The net result is that aldosterone synthase is ectopically expressed in the adrenal fasciculate under the control of ACTH rather than AII, the normal hormonal regulator. Aldosterone secretion thereby becomes linked to cortisol secretion such that maintenance of normal cortisol levels leads to constitutive aldosterone oversecretion, enhanced ENaC activity with increased distal tubular sodium reabsorption, volume expansion, and hypertension. The expanded plasma volume suppresses PRA but fails to suppress aldosterone production. Exogenous administration of glucocorticoid suppresses normal ACTH production and abrogates the ectopic production of aldosterone with normalization of BP.
There are several genetic disorders in which steroids other than aldosterone cause activation of the MR, leading to impaired natriuresis and salt-sensitive hypertension. The syndrome of apparent mineralocorticoid excess (AME) is an autosomal recessive disorder that causes early-onset hypertension with hypokalemic metabolic alkalosis (53). The PRA is suppressed and circulating aldosterone is absent. In normal individuals, circulating cortisol levels are 1,000-fold higher than aldosterone levels. In vitro, cortisol is known to bind and activate the MR. However, in vivo, virtually all MR activation is mediated by aldosterone. This paradox is explained by the finding that in the kidney MR specificity for aldosterone is mediated indirectly by the enzyme 11-β-hydroxysteroid dehydrogenase (11β-HSD). In the ENaC-containing principal cells of the collecting duct, expression of 11β-HSD results in metabolism of cortisol to cortisone, which is not capable of activating the MR. Thus, cortisol does not have a mineralocorticoid effect in normal individuals. However, in patients with AME, the absence of 11β-HSD allows cortisol to bind and activate the MR, resulting in salt-sensitive hypertension owing to ENaC-mediated enhancement of tubular sodium reabsorption. The development of hypertension following chronic ingestion of large amounts of natural licorice shares a similar pathogenesis. A licorice metabolite, glycyrrhetinic acid, is a potent inhibitor of 11β-HSD, resulting in a phenocopy of AME. Likewise, overproduction of cortisol caused by adrenal adenoma, ACTH-producing pituitary adenoma, or ectopic ACTH production, can overwhelm normal 11β-HSD activity so that cortisol is available for binding and activation of the MR, resulting in salt-sensitive hypertension with hypokalemic metabolic alkalosis.
Other genetic disorders are also associated with excess mineralocorticoid activity, resulting in chronic hypertension. In the adrenogenital syndromes, inherited autosomal recessive 11–b-hydroxylase deficiency (54) and 17a-hydroxylase deficiency (55) lead to impaired cortisol biosynthesis with compensatory hypersecretion of ACTH, which diverts steroid synthesis into pathways proximal to the enzymatic block. As a result, there is overproduction of 21-hydroxylated steroids such as deoxycorticosterone (DOC) and corticosterone, which are potent activators of the MR, leading to ENaC overexpression, enhanced sodium reabsorption in the DCT, and severe salt-sensitive hypertension (Fig. 9-1). The hypertension responds to cortisol replacement in these disorders, which suppresses ACTH and mineralocorticoid overproduction.
In contrast to disorders associated with excess mineralocorticoid that cause salt-sensitive hypertension, genetic disorders that impair aldosterone synthesis lead to mendelian forms of hypotension. Individuals homozygous for aldosterone synthase deficiency (16) have the phenotypic mirror image of GRA, with renal salt wasting and impaired secretion of K+ and H+ in the distal nephron (Fig. 9-1). These individuals present with severe hypotension caused by reduced intravascular volume with hyperkalemic metabolic acidosis. Likewise, homozygous 21-hydroxylase deficiency (56) results in the absence of circulating aldosterone, leading to volume depletion and hypotension (Fig. 9-1).
BLOOD PRESSURE VARIATIONS CAUSED BY MUTATION IN THE MINERALOCORTICOID RECEPTOR
A mutation in the ligand-binding domain of the MR causes an autosomal dominant form of hypertension that develops before the age of 20 years and increases markedly in severity during pregnancy (57). Carriers of this mineralocorticoid receptor missense mutation (MR S810L) develop hypertension at a young age. Steroids lacking 21-hydroxyl groups, such as progesterone, normally bind but fail to activate the MR. However, in carriers of the MR S810L missense mutation, progesterone binds and functions as a potent activator of MR. Because progesterone levels rise 100-fold during pregnancy, it is not surprising that all pregnancies among women harboring this mutation have been complicated by the development of severe pregnancy-induced hypertension accompanied by complete suppression of the RAAS.
In contrast to gain-of-function mutations of the MR, which cause hypertension, loss-of-function mutations in the MR cause renal salt wasting and hypotension. There are both autosomal dominant and autosomal recessive forms of the disease. Autosomal dominant pseudohypoaldosteronism type 1 (PHA1) is characterized by neonatal salt wasting with hypotension despite markedly elevated aldosterone levels in association with hyperkalemic metabolic acidosis (Fig. 9-1). Affected kindred have heterozygous loss-of-function mutation of the MR due to various premature terminations or frameshift mutations (58). The resulting partial loss of MR function impairs maximal sodium reabsorption, leading to salt wasting and hypotension. Insufficient ENaC activity causes a diminution in the electrical driving force for H+ and K+ secretion resulting in hyperkalemia and metabolic acidosis. In the neonatal period, two normal copies of the MR are apparently required for normal sodium homeostasis because with consumption of a normal salt-rich diet, older affected individuals become normotensive with resolution of the biochemical abnormalities.
BLOOD PRESSURE VARIATIONS CAUSED BY ALTERATIONS IN RENAL SODIUM CHANNELS AND TRANSPORTERS
Mutations leading to a gain-of-function mutation in the ENaC also cause salt-sensitive hypertension. Liddle syndrome is characterized by autosomal dominant transmission with early-onset hypertension in association with hypokalemic metabolic alkalosis, suppressed PRA, and low plasma aldosterone levels. This disease is caused by mutations in either the β or γ subunit of the ENaC in which there is deletion of their cytoplasmic carboxy termini (59). These mutations result in enhanced ENaC activity that is attributable to an increase in the number of ENaC inserted into the luminal membrane of principal cells in the DCT. The enhanced number of channels is caused by reduced clearance of ENaC from the cell membrane that substantially prolongs ENaC half-life. The increase in ENaC activity leads to enhanced sodium reabsorption, increase in net renal sodium balance, and salt-sensitive hypertension. The pivotal role of impaired natriuresis in the pathogenesis of hypertension is further illustrated by a case report in which severe hypertension in a patient with Liddle syndrome was cured by successful kidney transplantation from a normotensive donor (60). The most common ENaC variant is the T594M mutation, which causes a gain-of-function in the β-subunit, and has been found among some individuals with essential hypertension. In a case–control study, 206 hypertensive and 142 normotensive blacks who lived in London were screened for the T594M mutation. It was found that 17 (8.3%) of the hypertensive participants compared with 3 (2.1%) of the normotensive participants possessed the T594M variant (61). These findings suggested that the T594M mutation could contribute to secondary essential hypertension in black people.
In contrast to gain-of-function mutations in ENaC that cause hypertension, loss-of-function mutations result in hypotension. Autosomal recessive PHA1 is caused by loss-of-function mutation in any one of the three ENaC subunits (Fig. 9-1). This disorder causes life-threatening renal salt wasting and hypotension, with hyperkalemic metabolic acidosis despite elevated aldosterone levels (62). Like the autosomal dominant form of PHA1, this disorder begins in the neonatal period; however, it does not resolve on consumption of a salt-rich diet. Affected individuals require lifelong treatment with massive salt supplementation and treatment for hyperkalemia.
To date, hypertension resulting from gain-of-function mutations in the sodium transporters in the PT, TAL, or DCT has not been reported. However, loss-of-function mutations in the Na/K/2Cl cotransporter in the TAL and the thiazide-sensitive NCC in the DCT have been well characterized (16). These autosomal recessive disorders are associated with low–normal BP in association with hypokalemic metabolic alkalosis. In all cases, the disease is caused by mutations that result in renal sodium wasting. Affected individuals can present in the neonatal period with life-threatening hypotension owing to renal sodium wasting or can have disease that is found incidentally. The Gitelman syndrome results from homozygous loss-of-function mutations in the thiazide-sensitive NCC (Fig. 9-1) (63). Renal salt wasting leads to BP that is lower than in the general population. Patients present in adolescence with neuromuscular symptoms resulting from hypokalemia. Like thiazide diuretic-treated patients, they have hypomagnesemia and hypocalciuria. The salt wasting at the level of DCT leads to compensatory activation of the RAAS. Activation of the MR leads to augmentation of ENaC activity, thereby enhancing sodium reabsorption at the expense of increased H+ and K+ secretion leading to hypokalemic metabolic alkalosis. Linkage studies in a large Gitelman kindred indicate that loss-of-function mutations in the NCC indeed lower BP, thereby providing convincing evidence that even a modest enhancement of the natriuretic capacity of the kidney causes a reduction in BP (64). The Bartter syndrome occurs in individuals homozygous for various loss-of-function mutations in any of the three genes required for normal function of the Na/K/2Cl cotransporter in the TAL (Fig. 9-1) (16,65). Impaired function of the transporter leads to marked renal salt wasting, reflex activation of the RAAS, and hypokalemic metabolic alkalosis (Fig. 9-1) (16). In type I Bartter syndrome, the mutation may reside in the Na/K/2Cl cotransporter. In type II, the defect resides in the ATP-sensitive K+ channel ROMK, which is required for potassium recycling and efficient reabsorption of Na+ and K+ in the TAL. Type III is caused by homozygous mutation in the chloride channel (CLCNKB), which is required for chloride exit from the cell across the basolateral membrane (16). Individuals with Bartter syndrome often present with premature delivery and life-threatening hypotension caused by salt wasting in the neonatal period. In contrast to Gitelman syndrome, Bartter syndrome is associated with hypercalciuria and normal or only slightly reduced magnesium levels (similar to patients treated with loop diuretics).
Gain-of-function mutations in the sodium–hydrogen ion exchanger (NHE-3) in the PT have been investigated as a possible cause of essential hypertension in the general population; however, linkage analysis studies have failed to demonstrate an association (66).
IMPAIRED NATRIURESIS CAUSED BY HYPERINSULINEMIA
It has been observed that glucose intolerance, hyperlipidemia, and essential hypertension tend to cluster in the same patients. In fact, essential hypertension often occurs many years before the onset of overt diabetes with hyperglycemia. It has been proposed that insulin resistance is central to the pathogenesis of this so-called “syndrome X,” a condition now known as metabolic syndrome (67). Insulin resistance, which may be inherited or acquired (owing to obesity, dietary factors, or sedentary life style), results in compensatory hyperinsulinemia. Eventually, the β-cell output of insulin may become inadequate to compensate for insulin resistance, resulting in glucose intolerance or frank type 2 diabetes. The hyperinsulinemia induces abnormalities of lipid metabolism with increased low-density lipoprotein and reduced HDL levels. Hyperinsulinemia also may be causally related to the development of hypertension. Insulin has been shown to play an important role in renal sodium handling. In human studies in which euglycemic hyperinsulinemia was generated using an insulin clamp technique, urinary sodium excretion declined significantly within 60 minutes (68). This antinatriuretic effect of insulin was observed in the absence of changes in glomerular filtration rate (GFR), renal plasma flow, the filtered load of glucose, or plasma aldosterone concentration (PAC). The predominant effect of insulin on tubular sodium reabsorption is in more distal parts of the nephron (TAL of Henle or DCT). Thus, the net effect of insulin resistance and resulting hyperinsulinemia is to induce an impairment in the intrinsic natriuretic capacity of the kidney, which results in the development of salt-sensitive hypertension.
ACQUIRED TUBULOINTERSTITIAL DISEASE IN THE PATHOGENESIS OF SALT-DEPENDENT HYPERTENSION
There is compelling evidence that essential hypertension results from a defect whereby the kidney is unable to maintain sodium homeostasis at a normal BP. However, if the defect that causes impaired natriuresis is genetic, then why does hypertension generally not develop until adulthood? The salt-sensitive hypertension associated with obesity and aging appears to be an acquired disorder. Why does salt sensitivity increase in frequency and magnitude over time? It has been proposed that salt-sensitive hypertension may be the result of acquired tubulointerstitial renal disease (69). The hypothesis is that hypertension consists of two phases. The first phase is characterized by episodic elevations in BP caused by hyperactivity of the sympathetic nervous system (elevated plasma norepinephrine [NE] and baroreceptor sensitivity) induced by genetic or environmental factors. Less commonly, the episodic hypertension relates to activation of the renin–angiotensin system (RAAS). Elevations of BP are episodic and renal sodium handling is normal during this initial phase. Transition to the second phase is proposed to occur as a consequence of transient catecholamine or AII-mediated elevations in BP that preferentially damage the juxtamedullary and medullary regions, which do not autoregulate, as well as cortical regions in response to sudden changes in renal perfusion pressure. The pressor response may be associated with both an increase in peritubular capillary pressure and a reduction in peritubular capillary flow, resulting in injury to the peritubular capillaries with ischemia of the tubules and interstitium. The resulting tubulointerstitial injury then may trigger local vasoconstrictor mechanisms (AII, adenosine, or renal sympathetic nerves) or inhibit vasodilator mechanisms (nitric oxide [NO], prostaglandins, or dopamine), further augmenting ischemia and resulting in abnormal tubuloglomerular feedback and enhanced tubular sodium reabsorption. Peritubular capillary damage and rarefaction lead to an increase in renovascular resistance, which further blunts the pressure natriuresis mechanism. The predicted consequence of enhanced tubuloglomerular feedback and impaired pressure natriuresis is an acquired functional defect in renal sodium excretion. This resetting of the pressure natriuresis curve to higher pressure is proposed to be the explanation for the development of acquired salt-sensitive hypertension.
Animal models confirm that short-term exposure to catecholamines or AII causes renal injury, which leads to the development of salt-sensitive hypertension that persists even after catecholamine and AII infusions are stopped (70,71). An 8-week infusion of phenylephrine by mini pump was found to induce structural and functional changes in the kidneys of rats (70). Glomeruli were spared but focal tubulointerstitial fibrosis was present in association increased expression of transforming growth factor-β (TGF-β), de novo expression of the macrophage adhesive protein osteopontin by injured tubules, macrophage and α-smooth muscle actin-positive myofibroblast accumulation in the interstitium, as well as distortion and rarefaction of the peritubular capillaries. BP returned to normal in rats maintained on low-salt diet after discontinuation of catecholamine infusion. However, rats fed on a high-salt diet developed marked hypertension. Short-term (2-week) infusion of AII also caused sustained salt-sensitive hypertension in association with tubulointerstitial damage and fibrosis (71).
This hypothesis may help to explain the development of salt-sensitive hypertension in some high-risk populations. For instance, the prevalence of salt-sensitive hypertension increases progressively with age in the general population. Aging is associated with a progressive decline in renal function and the development of glomerulosclerosis and interstitial fibrosis. In rat models, aging is associated with tubulointerstitial fibrosis characterized by tubular injury, myofibroblast proliferation, osteopontin expression, macrophage infiltration, and collagen IV deposition (72). These structural changes could lead to impaired natriuresis with the development of salt-sensitive hypertension. Obesity also is associated with an increased prevalence of hypertension. Obese individuals have been shown to have augmented sympathetic nervous system activity with higher basal NE and plasma renin levels, glomerular hyperfiltration, increased prevalence of interstitial fibrosis and glomerulosclerosis, expanded ECF volume, and salt-dependent hypertension (69). In transplant patients, treatment with cyclosporine A (CsA) to prevent allograft rejection is also associated with interstitial fibrosis, osteopontin and TGF-β expression, and the development of salt-sensitive hypertension in humans (69). Cyclosporine is a vasoconstrictor that can directly inhibit NO production. Rats administered CsA develop interstitial fibrosis identical to that observed in humans with tubulointerstitial injury preferentially involving the juxtaglomerular regions. Interstitial fibrosis develops in association with TGF-β and osteopontin expression and reduction in endothelial NO synthase. If CsA administration is stopped after the tubulointerstitial lesion develops, placement of the animals on a high-salt diet results in rapid development of hypertension despite the presence of normal GFR (69).
IMPAIRED NATRIURESIS CAUSED BY REDUCED NEPHRON MASS
It has been postulated that the underlying cause of impaired natriuretic capacity in essential (genetic) hypertension may be a congenitally acquired deficit of effective nephron mass (29). In the SHR, the fenestrae of the glomerular capillary endothelium are smaller than in normotensive control rats. In the Milan SHR, there is a decreased glomerular ultrafiltration coefficient and increased proximal sodium reabsorption. Salt-sensitive hypertension in rats is associated with a 15% reduction in nephron number. In humans, major inborn deficits of nephron number such as oligomeganephronia and congenital unilateral renal agenesis are associated with the development of hypertension. Brenner has postulated that the abnormality that predisposes a minority of the population to essential hypertension in the setting of excessive sodium intake is an inherited deficit of nephrons or glomerular filtration surface area leading to a diminished capacity to excrete a sodium load resulting in salt-sensitive hypertension (29). Of course, CKD of any etiology could also lead to a state of impaired natriuretic capacity with resulting salt-sensitive hypertension.
IMPAIRED NATRIURESIS CAUSED BY ABNORMAL REGULATION OF THE RENIN–ANGIOTENSIN SYSTEM
Increased circulating levels of AII stimulate increased production of aldosterone with activation of the MR and enhanced sodium reabsorption in the distal nephron as discussed in detail in the preceding section. AII has also been shown to increase renal sodium and water retention independent of its effect to increase aldosterone production (73). In this regard, AII-mediated renal hemodynamic changes lead indirectly to increases in tubular sodium reabsorption. Infusion of AII causes a reduction in renal blood flow with maintenance of near-normal GFR because of an increase in filtration fraction consistent with an increase in efferent arteriolar resistance. The increase in efferent arteriolar resistance caused by AII results in a fall in hydrostatic pressure and an increase in oncotic pressure in the peritubular capillaries of the proximal and distal tubules and collecting ducts, which results in enhanced tubular sodium reabsorption (73). AII also has been shown to directly stimulate sodium reabsorption in the PT (74).
Recent evidence suggests that renal abnormalities involving disordered regulation of the RAAS may be important in the pathogenesis of essential hypertension (75). These fundamental abnormalities, which are present in 45% of patients with essential hypertension, cause impairment in renal sodium handling resulting in sodium-sensitive hypertension. These patients have normal to high PRA and have been termed “nonmodulators” because of their inability to appropriately adjust activity of the RAAS during changes in sodium intake. Renal blood flow changes in parallel to sodium intake in normal individuals or patients with essential hypertension and who are “modulators.” Renal blood flow rises with an increase in sodium intake, whereas renal blood flow declines with a decrease in sodium intake. In contrast, the renal blood flow remains fixed in nonmodulators despite changes in sodium intake. This abnormal renal vascular response to changes in sodium intake may account for the limited capacity of the kidney to handle a sodium load. Nonmodulators demonstrate less suppression of renin in response to sodium loading or AII infusion compared with normal individuals or modulators with essential hypertension. Treatment of these nonmodulators with an ACE inhibitor restores the normal natriuretic response to a sodium load and normalizes renin suppression in response to a sodium load or AII infusion. Furthermore, ACE inhibition also results in a decrease in renal vascular resistance and an increase in renal blood flow and natriuresis (75). These findings suggest that enhanced renal vascular responsiveness to AII is present in nonmodulators, even in the absence of increased systemic renin and AII levels, perhaps secondary to in situ AII production by renal-converting enzyme. The resulting decrease in peritubular Starling force and increase in proximal and distal sodium reabsorption may underlie the defect in natriuresis in some cases of essential hypertension. Moreover, restoration of the ability of the kidney to handle a sodium load may explain why treatment with ACE inhibitors leads to a reduction in BP in patients with low-renin essential hypertension.
SYMPATHETIC NERVOUS SYSTEM-MEDIATED IMPAIRMENT IN NATRIURESIS
Neural mechanisms activated in response to perceived changes in BP or intravascular volume act on the kidney to modulate renal sodium excretion. Proximal tubular sodium reabsorption is enhanced by α-adrenergic receptor-mediated sympathetic activation (76). Increased vasoconstrictor responsiveness of the efferent arteriole to α-adrenergic stimuli may cause the intrarenal hemodynamic abnormalities typical of patients with essential hypertension, namely decreased renal blood flow, increased renal vascular resistance, and increased filtration fraction (77). Akin to the effect of AII, this primary increase in efferent resistance may contribute to the defective natriuresis in essential hypertension by altering peritubular capillary Starling forces. In addition, increased α-adrenergic activity has been shown to directly stimulate sodium reabsorption in the PT (78). Calcium channel blockers (CCBs) decrease renal efferent arteriolar vasoconstriction and improve renal blood flow (77). The natriuretic response to CCBs may be secondary to this phenomenon in addition to their direct effect on tubular sodium transport (79).
The renal nerves contribute to development of hypertension in experimental models, and appear to play a role in the pathogenesis of hypertension in humans (80). In both the SHR and DOC-salt rat models of hypertension, the full development of hypertension is dependent on renal afferent nerve activity, which stimulates renal tubular sodium reabsorption. Denervation of the kidney ameliorates hypertension in these models (80).
IMPAIRED NATRIURESIS CAUSED BY ABNORMALITIES IN RENAL NITRIC OXIDE
There is increasing evidence that endothelium-derived NO is tonically synthesized within the kidney and that NO plays a crucial role in the regulation of renal hemodynamics and sodium excretion (81). These effects are mediated in part by interactions between NO and the RAAS. NO is an important mediator of renal blood flow and the renal microcirculation. Bradykinin and acetylcholine induce renal vasodilation by increasing NO synthesis, which in turn leads to enhancement of diuresis and natriuresis. Blockade of basal NO synthesis, with specific inhibitors of the L-arginine NO pathway (L-NAME or L-NMMA), has been shown to result in an increase in renal vascular resistance with decreases in renal blood flow, urine flow, and sodium excretion. Intrarenal inhibition of NO synthesis leads to a reduction in sodium excretory responses to changes in renal perfusion pressure without an effect on renal autoregulation, suggesting that NO exerts a permissive or mediatory role in the tubular responses that regulate the pressure natriuresis mechanism (82,83). NO released from the macula densa may modulate tubuloglomerular feedback response by affecting afferent arteriolar constriction. In the collecting duct, an NO-dependent inhibition of solute transport has been suggested. Although most studies indicate that NO synthesis blockade causes reduction in sodium excretion, the exact mechanism is unclear. Reduction in the filtered load of sodium, increased tubular sodium reabsorption, altered medullary blood flow, and decreased renal interstitial hydrostatic pressure may mediate sodium retention during NO blockade. Taken together, these observations suggest that NO-dependent mechanisms have a major impact on renal volume control. Abnormalities in renal NO-mediated sodium excretion have been postulated to cause a state of impaired natriuresis, resulting in salt-sensitive hypertension (81–83). In this regard, impaired NO synthase activity related to insulin resistance may contribute to the pathogenesis of salt-sensitive hypertension in patients with the metabolic syndrome (84).
IMPAIRED NATRIURESIS CAUSED BY ENHANCED PROXIMAL TUBULAR SODIUM REABSORPTION
In some cases, the genetic defect that underlies the development of essential hypertension may be a diminished natriuretic capacity caused by a primary increase in PT sodium reabsorption (85). Lithium clearance often is used as a surrogate marker of proximal tubular sodium reabsorption because lithium reabsorption occurs in parallel to sodium resorption and is limited to the PT. Therefore, a decreased fractional lithium clearance (ratio of lithium clearance to creatinine clearance) implies increased proximal sodium reabsorption. Studies in patients with essential hypertension have revealed a decreased fractional lithium clearance (85). Furthermore, normotensive subjects with a hypertensive first-degree relative had a lower fractional lithium clearance than subjects with no hypertensive relative (85).
IMPAIRED NATRIURESIS: A PREREQUISITE IN HYPERTENSION
Experiments with isolated, perfused kidneys demonstrate that the magnitude of urinary sodium excretion is a direct function of the perfusion pressure (86). The level of perfusion pressure may alter sodium excretion by changing the peritubular hydrostatic pressure. Thus, an increase in perfusion pressure should increase peritubular hydrostatic pressure with a resultant decrease in sodium reabsorption. Micropuncture studies in the rat have shown an inverse relationship between renal perfusion pressure and proximal sodium reabsorption (87).
It has been argued that if this pressure natriuresis mechanism were operating in a normal fashion, then profound volume depletion would occur in the setting of hypertension. The fact that this does not occur suggests that in every hypertensive state, there must be a shift in the pressure natriuresis curve such that a higher perfusion pressure is required to achieve any given level of natriuresis. In this regard, it has been postulated that this shift in the pressure natriuresis curve is actually a reflection of the underlying renal abnormality present in essential hypertension as well as in hypertension caused by CKD (21,24,25). If a primary defect in natriuresis does exist in hypertension, then to avert disaster owing to persistent positive sodium balance with inexorable fluid accumulation, compensatory hormonal responses or other mechanisms must be invoked that restore sodium balance. The theories regarding the pathogenesis of hypertension that follow explain how these compensatory processes restore sodium balance but in the process cause systemic hypertension in association with an elevated systemic vascular resistance.
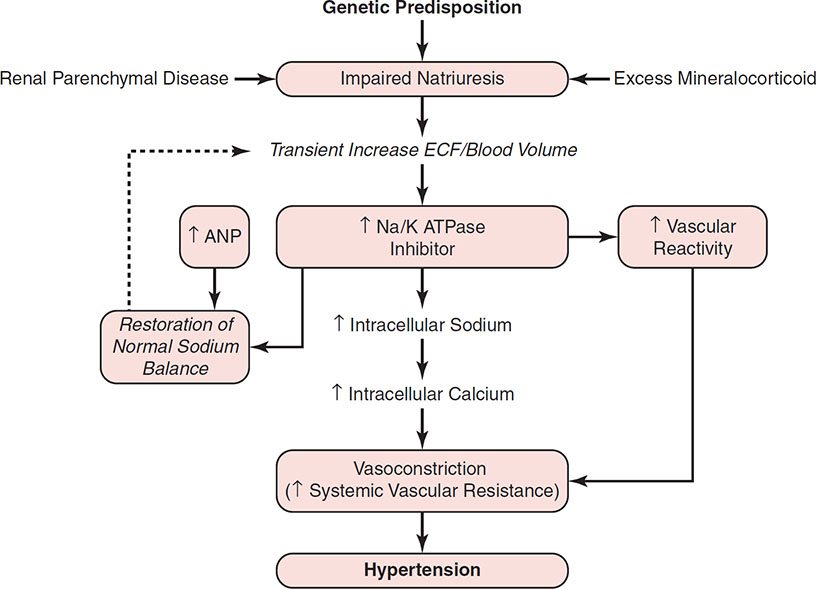
The Na/K ATPase Inhibitor Hypothesis
There must be an underlying abnormality in the kidney’s ability to excrete sodium in both essential hypertension and secondary hypertension caused by renal disease. In individuals with this impairment of natriuretic capacity, if the intake of dietary sodium is >60 mmol/day, then there will be a tendency toward sodium and water retention resulting in ECF volume expansion. Some authors have proposed that in response to this expansion of ECF volume, there is an increase in the plasma concentration of two or more substances, collectively referred to as “natriuretic hormone” (21,22,88,89). The responses induced by this natriuretic hormone include an increase in the natriuretic capacity of the plasma, an increase in the ability of the plasma to inhibit Na/K ATPase, and an increase in vascular responsiveness to vasoconstrictors such as NE, AII, and vasopressin. Atrial natriuretic peptide (ANP), which is released from the atria in response to acute volume expansion, causes a brisk increase in renal sodium and water excretion of rapid onset and short duration. However, ANP does not inhibit Na/K ATPase or increase vascular reactivity; in fact, ANP decreases systemic vascular resistance. It is proposed that another response to the underlying renal impairment in the ability to excrete sodium is an increase in the plasma concentration of a substance, probably of hypothalamic origin, which inhibits Na/K ATPase (90,91). As a result of inhibition of Na/K ATPase, renal tubular sodium reabsorption is reduced and urinary sodium excretion increases, thereby returning sodium balance toward normal. However, this circulating inhibitor also inhibits the sodium pump in other cells, such as erythrocytes and leukocytes and, more importantly, in vascular smooth muscle cells. The increase in cellular sodium is associated with increased Na/Ca exchange and increased cellular calcium concentration. Thus, at the arteriolar level, inhibition of Na/K ATPase theoretically could cause vasoconstriction secondary to increased intracellular calcium with a resultant increase in systemic vascular resistance and a rise in BP (88). As a result of the compensatory release of these natriuretic substances, sodium balance and ECF volume are restored to normal but at the expense of systemic hypertension caused by the increase in systemic vascular resistance (Fig. 9-2). Thus, although the underlying cause of this type of salt-sensitive (volume-dependent) hypertension is a defect in renal natriuretic capacity, this does not result in a detectable increase in ECF volume or cardiac output in the steady-state phase. Instead, the hypertension is maintained by the resulting increase in systemic vascular resistance (21,22,88,89).
The Guyton Hypothesis
Guyton’s hypothesis is that the most important and fundamental mechanism determining the long-term control of BP is the renal fluid–volume feedback mechanism. In simple terms, this is the basic mechanism through which the kidneys regulate arterial pressure by altering renal excretion of sodium and water, thereby controlling circulatory volume and cardiac output. Changes in BP, in turn, directly influence renal excretion of sodium and water, thereby providing a negative feedback mechanism for control of ECF volume, cardiac output, and BP. The hypothesis is that derangements in this renal fluid–volume pressure control mechanism are the fundamental cause of virtually all hypertensive states (21,23–25,92–94).
RENAL FUNCTION CURVES
Interactions of the renal perfusion pressure–sodium excretion mechanism and modulating neurohormonal factors normally operate very precisely to maintain sodium balance at a normal arterial pressure (92). The physiologic basis of the renal body fluid feedback mechanism for the regulation of arterial pressure is the direct effect of arterial pressure on output of water and sodium from the kidneys. Studies of the isolated, perfused kidney demonstrate the so-called pressure natriuresis and diuresis whereby an increase in perfusion pressure directly causes the renal output of sodium and water to increase (93,94). Figure 9-3 depicts a renal function curve that shows the effect of perfusion pressure on urinary sodium excretion in the isolated perfused kidney. Urinary sodium output falls to zero when the arterial pressure falls to approximately 50 mm Hg. In contrast, the output of sodium increases sixfold to eightfold when the arterial pressure rises from the normal value of 100 to 200 mm Hg (92). This effect of arterial pressure on sodium excretion has been demonstrated in isolated, perfused kidneys and in intact animals. However, for the reasons discussed in the following, the upward slope of the renal function curve in the intact animal is much steeper. The horizontal line in Figure 9-3 represents the level of sodium intake at equilibrium when sodium intake and output are matched. When net intake and output of sodium are matched, the arterial pressure is determined by the point where the two plots intersect, which is known as the equilibrium pressure point. Computer model analysis of hypothetical renal function curves in the intact animal suggests that, if the renal function curve and the sodium intake remain unchanged, this is the unique perfusion pressure at which external sodium balance will be maintained (95). If the pressure rises above this level, the sodium output becomes greater than input and negative sodium balance occurs with eventual reduction in ECF volume and cardiac output to a level that returns the BP to the equilibrium point. In contrast, if the BP falls below the equilibrium point, the intake of sodium will be greater than the output and a positive sodium balance will occur until the increases in ECF volume, blood volume, and cardiac output are sufficient to return the pressure to the equilibrium point. Sodium balance can be maintained only at the 100 mm Hg equilibrium pressure point.

Figure 9-3 Renal function curves showing the effect of renal perfusion pressure on urinary sodium excretion in the isolated, perfused kidney. By using computer modeling, the corresponding sodium intake can be superimposed on the pressure natriuresis curve. An equilibrium pressure point is defined, which represents the unique level at which the arterial pressure will be regulated. It is apparent that a primary increase in systemic vascular resistance alone cannot lead to sustained increase in blood pressure (BP) because the pressure natriuresis mechanism would cause sodium excretion, decreased extracellular fluid (ECF) volume, and cardiac output, thereby returning the BP to the equilibrium point. Likewise, a primary decrease in systemic vascular resistance would not lead to sustained hypotension because reduced natriuresis, sodium retention, increased ECF volume, and cardiac output would once again return BP to the equilibrium point. (From Guyton AC. Renal function curve: a key to understanding the pathogenesis of hypertension. Hypertension. 1987;10:1, with permission from Wolters Kluwer Health, Inc.)
If this model, illustrated in Figure 9-3, is correct, then it is apparent that a primary increase in cardiac output or systemic vascular resistance cannot result in a sustained increase in BP because a normally functioning feedback mechanism would result in natriuresis and diuresis, thereby returning the BP to normal. Thus, a primary increase in systemic vascular resistance would be accompanied by an equal and opposite decrease in cardiac output with return of the BP to normal. This return of the pressure to the equilibrium point illustrates the infinite gain characteristic of the renal fluid–volume feedback mechanism. In this system, a change in the arterial pressure is the critical feedback stimulus that modifies the natriuretic response. In theory, if an initial decrease in BP results from a decrease in cardiac output, for instance in the setting of congestive heart failure, then the compensatory salt and water retention would increase ECF volume but fail to normalize cardiac output and renal perfusion pressure. Thus, renal sodium and water retention would continue unopposed, resulting in massive fluid overload. This mechanism is consistent with the unifying hypothesis recently proposed to explain body fluid volume regulation in disorders characterized by low effective arterial blood volume (28,96). The implication of this renal fluid–volume feedback mechanism is that the finding of sustained hypertension must be a reflection of an underlying abnormality that caused a shift of the renal function curve to the right such that a higher BP is required to maintain sodium balance at any given level of sodium intake (97–99).
ROLE OF THE RENIN–ANGIOTENSIN SYSTEM IN REGULATION OF BLOOD PRESSURE
Unlike the isolated, perfused kidney or computer models, in vivo the position of the renal function curve can be shifted by various neural and endocrine factors. For example, changes in the activity of the RAAS can result in a shift of the curve and thus either magnify or blunt the basic relation between sodium and water excretion and BP (95,98). Different renal function curves can be produced in animals by varying sodium intake in stepwise increments while maintaining AII levels constant at various levels by using combinations of ACE inhibitor and AII infusion (Fig. 9-4) (97). With the AII level maintained above normal, there is a shift in the renal function curve to the right consistent with a blunting of the pressure-induced natriuretic response. In contrast, when AII is totally suppressed by an ACE inhibitor, the curve is shifted to the left, consistent with an exaggerated pressure natriuresis. The vertical line in Figure 9-4 represents a different type of renal function curve called a salt-loading renal function curve. It is obtained when sodium intake is varied in a stepwise fashion in animals with an intact RAAS (AII level allowed to vary in response to the sodium intake), which will modulate the intrinsic renal body fluid feedback mechanism. Thus, the renal function curve in the intact animal is much steeper than that seen in the isolated perfused kidney. In this salt-loading curve, BP at equilibrium for each level of sodium intake changes very little. Analysis of the superimposed renal function curves, with AII held constant at different levels, illustrates that the steepness of the curve in the intact animal may be owing to changes in the activity of the RAAS. A high sodium intake suppresses the RAAS and shifts the renal function curve to the left, whereas a low sodium intake activates the RAAS and shifts the curve to the right. This modulation of the renal function curve by the RAAS is thought to result from the effect of AII on renal sodium and water reabsorption. AII directly enhances proximal tubular sodium reabsorption (74). In addition, AII has important renal hemodynamic effects that cause increased tubular sodium reabsorption independent of aldosterone (73). The predominant efferent vasoconstriction produced by AII causes a drop in peritubular capillary hydrostatic pressure, leading to enhanced tubular reabsorption of sodium and water (73,99). This dynamic interaction between the RAAS and renal fluid–volume feedback mechanism accounts for the observation that tremendous extremes of dietary sodium intake in normal individuals result in relatively little change in the systemic arterial pressure.

Figure 9-4 Modulation of renal function curves by the renin–angiotensin system. A salt-loading renal function curve is obtained when sodium intake is varied in stepwise increments in animals with an intact renin–angiotensin system. The curve is extremely steep such that over a wide range of dietary sodium intake, the blood pressure (BP) changes very little. Three separate renal function curves were produced by varying the sodium intake in a stepwise fashion in animals, with angiotensin II (AII) levels maintained constant in either the normal range, 2.5 times normal, or with AII absent. The curve obtained when AII production is suppressed by angiotensin-converting enzyme inhibitor is shifted to the left, consistent with an enhanced natriuretic response to any given level of perfusion pressure. In contrast, with an AII level at 2.5 times normal, the curve is shifted to the right, reflecting a blunting of the intrinsic renal natriuretic response. Superimposition of these individual renal function curves reveals that the steepness of the salt-loading renal function curve in the intact animal is caused by changes in the natriuretic response to pressure, which are in turn mediated by changes in the activity of the renin–angiotensin system as a function of dietary sodium intake. AII is suppressed with high sodium intake, producing a shift of the renal function curve to the left so the salt load can be excreted at normal BP. In contrast, AII production is enhanced with dietary sodium restriction, leading to a shift of the curve to the right such that the kidney is more avid for sodium such that a normal level of BP is still maintained. (From Guyton AC. Renal function curve: a key to understanding hypertension. Hypertension. 1987;10:1, with permission from Wolters Kluwer Health, Inc.)
AUTOREGULATION LEADS TO INCREASED SYSTEMIC VASCULAR RESISTANCE
The ability to maintain normal BP over a wide range of dietary sodium intake is present only if both the RAA system and the kidney function normally. Aberrations in either the RAA system or the renal fluid–volume mechanism can lead to a significant increase in BP when the sodium intake is increased. Guyton’s hypothesis holds that there are two basic ways in which the pressure equilibrium set point can be increased with resulting hypertension. A shift of the renal function curve to the right along the pressure axis owing to either intrinsic renal abnormalities or overactivity of the RAA system can cause hypertension. Alternatively, an increase in the sodium intake without a compensatory leftward shift of the renal function curve also can cause hypertension.
In the setting of an underlying decrease in the inherent natriuretic capacity, the renal fluid–volume feedback mechanism should cause progressive sodium and water retention until the increases in ECF volume, blood volume, and cardiac output are sufficient to raise the BP to the equilibrium pressure point. Sodium and water balance is restored at the equilibrium point; however, this is accomplished at the expense of systemic hypertension (Fig. 9-5). Thus, when the inherent natriuretic capacity is reduced, Guyton’s concept holds that an increase in arterial pressure is an essential protective mechanism to restore sodium balance and avert disaster (21–25,94).
Theoretically, hypertension caused by a rightward shift of the renal function curve should be mediated by an increased cardiac output in response to sodium and water retention. However, in animal models and humans with essential hypertension, even though the initiating mechanism for hypertension is sodium retention with increased ECF volume and cardiac output, ultimately an increase in the peripheral vascular resistance perpetuates the hypertension, whereas the cardiac output and ECF volume return to normal. According to Guyton’s hypothesis, this transition from high cardiac output hypertension to the high systemic vascular resistance type of hypertension is explained by the process of autoregulation of blood flow in the systemic circulation (23–25). Autoregulation is a local tissue phenomenon that adjusts local blood flow when it becomes too high or low. Acutely, autoregulation may result from local changes in vascular muscle tone; however, structural changes in the resistance vessels develop in the chronic phase (18). In theory, when the cardiac output increases as a result of ECF volume expansion, autoregulatory vasoconstriction in all vascular beds eventually returns the cardiac output to normal. Hypertension persists, however, because the fall in cardiac output is accompanied by an equal and opposite increase in systemic vascular resistance. Given the persistent hypertension, sodium balance can still be maintained by the renal fluid–volume pressure natriuresis mechanism. Figure 9-5 summarizes the pathophysiologic sequence whereby the various disorders that cause an initial defect in natriuretic capacity (shift of renal function curve to the right) eventually lead to sustained hypertension caused by increased systemic vascular resistance in the absence of clinically evident increases in ECF volume, blood volume, or cardiac output.
Related posts:
 Disorders of Water Homeostasis
Disorders of Water Homeostasis
 Pathogenesis and Management of Metabolic Acidosis and Alkalosis
Pathogenesis and Management of Metabolic Acidosis and Alkalosis
 Renal Physiology and Pathophysiology in Pregnancy
Renal Physiology and Pathophysiology in Pregnancy
 Acute Kidney Injury: Pathogenesis, Diagnosis, and Management
Acute Kidney Injury: Pathogenesis, Diagnosis, and Management
 Obstructive Nephropathy: Pathophysiology and Management
Obstructive Nephropathy: Pathophysiology and Management
 Pathophysiology and Management of Respiratory and Mixed
Acid–Base Disorders
Pathophysiology and Management of Respiratory and Mixed
Acid–Base Disorders
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


