Key points
- •
Multiple endogenous causes of hypogonadism may adversely affect a man’s fertility potential.
- •
Fertility potential in the hypogonadal male may be reestablished through medical or surgical therapies, depending on the pathology.
- •
Exogenous testosterone is a known and common cause of infertility in men.
- •
Exogenous testosterone should be discontinued in any male trying to achieve pregnancy. Supplemental therapy to stimulate spermatogenesis may be necessary.
Introduction
Infertility is the inability to conceive after 12 months of unprotected intercourse. Approximately 15% of couples have infertility, but only 1 in 5 of these couples seek evaluation and treatment. In 30% of infertile couples, a significant male factor alone is found as the cause for infertility. An additional 20% of couples have both male and female factors; therefore, male factors contribute to one-half of all infertile relationships. Endocrinopathies, including hypogonadism, are a common underlying problem in the infertile male, representing 9.6% of men presenting for infertility evaluation. The many endogenous causes of hypogonadism are listed in Box 1 and discussed in this paper. Exogenous testosterone, either prescribed by a physician or acquired by the patient elsewhere, is another commonly encountered cause of infertility. This paper discusses both exogenous testosterone and endogenous causes of hypogonadism as they relate to male infertility.
Klinefelter syndrome
Hyperprolactinemia
Hyperestrogenenemia
Congenital adrenal hyperplasia
Kallmann syndrome
Prader–Willi syndrome
Idiopathic hypogonadism
Introduction
Infertility is the inability to conceive after 12 months of unprotected intercourse. Approximately 15% of couples have infertility, but only 1 in 5 of these couples seek evaluation and treatment. In 30% of infertile couples, a significant male factor alone is found as the cause for infertility. An additional 20% of couples have both male and female factors; therefore, male factors contribute to one-half of all infertile relationships. Endocrinopathies, including hypogonadism, are a common underlying problem in the infertile male, representing 9.6% of men presenting for infertility evaluation. The many endogenous causes of hypogonadism are listed in Box 1 and discussed in this paper. Exogenous testosterone, either prescribed by a physician or acquired by the patient elsewhere, is another commonly encountered cause of infertility. This paper discusses both exogenous testosterone and endogenous causes of hypogonadism as they relate to male infertility.
Klinefelter syndrome
Hyperprolactinemia
Hyperestrogenenemia
Congenital adrenal hyperplasia
Kallmann syndrome
Prader–Willi syndrome
Idiopathic hypogonadism
Causes of hypogonadism
Male patients presenting with infertility and no history of exogenous testosterone use may have an underlying endocrinopathy resulting in hypogonadism (low testosterone). The American Urological Association Best Practice Statement recommends testing serum testosterone and follicle-stimulating hormone (FSH) if the patient has oligozoospermia (especially if the sperm concentration is <10 million/mL), impaired sexual function, or if the patient has other clinical findings suggestive of an endocrinopathy. Some physicians advocate a more extensive initial hormonal panel to include serum estradiol, luteinizing hormone (LH), sex hormone binding globulin, albumin, and/or prolactin.
The presence of testosterone is critical for spermatogenesis. Normal intratesticular testosterone levels are 50 to 100 times the concentration of testosterone found in serum. The interference with this high level of intratesticular testosterone is one of the reasons exogenous testosterone can either greatly impair or shut down spermatogenesis entirely ( Fig. 1 ). Likewise, endogenous hypogonadal states can be a cause for impaired spermatogenesis, and patients with hypogonadism can present with oligozoospermia or azoospermia.
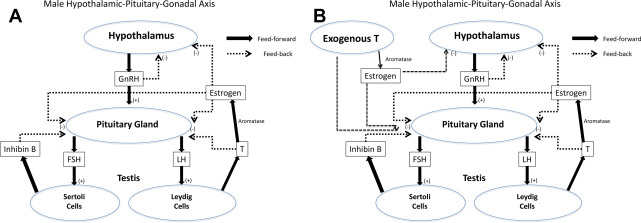
Hypogonadism is categorized as being either primary or secondary. Primary hypogonadism is failure at the level of the testis that is manifested by low serum testosterone and elevated gonadotropins (LH and FSH). Primary hypogonadism is also known as hypergonadotropic hypogonadism or primary testicular failure. Secondary hypogonadism, on the other hand, is hypogonadism in the setting of low or low normal gonadotropins, which is aptly named hypogonadotropic hypogonadism (HH).
Hypogonadotropic Hypogonadism
HH can be either congenital or owing to a variety of conditions that impair the hypothalamus or pituitary from properly releasing important trophic hormones for the testis. The hypothalamus releases gonadotropin-releasing hormone (GnRH) in pulsatile fashion to stimulate pituitary gonadotropes, which produce LH and FSH. Diseases or conditions that affect the pituitary and/or hypothalamus, including tumors, trauma, autoimmune disease, infarction, or infection, can disrupt gonadotropic function and lead to HH.
Kallmann syndrome
The most common congenital form of HH results from the failure of GnRH-releasing neurons to migrate to the olfactory lobe during development. An associated failure in development of the olfactory lobe leads to anosmia. Congenital anosmia and HH are the hallmarks of Kallmann syndrome, a cause of male infertility and, more rarely, female infertility, that occurs in 1 in 10,000 to 60,000 live births. Patients commonly present after failing to undergo puberty, but may also have midline facial defects, congenital deafness, cranial asymmetry, cryptorchidism, or renal abnormalities.
Multiple genetic mutations have been implicated in cases of Kallmann syndrome, which lead to a variety of inheritance patterns. These gene mutations include those in the X-linked KAL1 gene and the autosomal-dominant fibroblast growth factor receptor 1 gene, known as FGFR1 or KAL2 . An additional gene coding for a G-protein–coupled receptor, PROKR2 or PROK2 , have been found with apparent loss-of-function mutations leading to rare sporadic cases of Kallmann syndrome. These known gene mutations have only been found in fewer than one-third of cases, which underscores the need for further study of this rare condition.
Treatment of the delayed puberty and infertility associated with Kallmann syndrome is achieved by coordinated administration of gonadotropins. Testosterone therapy is used to initiate puberty in these patients, and, later, human chorionic gonadotropin (hCG) and human menopause gonadotropin (hMG) are used to induce spermatogenesis. With treatment, these patients can undergo adequate virilization and bone growth and many achieve pregnancy without the need for assisted reproductive technologies.
Prader–Willi syndrome
Prader–Willi Syndrome affects approximately 1 in 15,000 to 1 in 30,000 individuals and is characterized by excessive appetite, obesity, mental retardation, cryptorchidism, infantile hypotonia, and HH. It is most commonly caused by mutations or deletions of the paternally derived chromosome 15 at q11 or q13. It is uncommon for these patients to seek infertility treatment.
Other genetic causes
A variety of genetic defects have been linked to HH. An X-linked gene, DAX1 , is related to hypothalamic, pituitary, adrenal, and gonadal development, as well as maintaining spermatogenesis. DAX1 mutations can cause HH as well as congenital adrenal hyperplasia. In patients with HH, numerous loss-of-function mutations have been identified in GnRHR , the gene that encodes the GnRH receptor found on pituitary gonadotropes. Additionally, mutations have been found in the gene that encodes LH as well as the LH receptor, both of which have been found to cause hypogonadism in subfertile males.
Hyperprolactinemia
The presence of excess serum prolactin inhibits LH action on Leydig cells, thereby causing hypogonadism. According to the American Urological Association Best Practice Statement, a serum prolactin measurement should be obtained if the patient’s testosterone level is found to be low. Hyperprolactinemia may be owing to a pituitary adenoma and warrants further workup. A careful history and physical examination should include questions about headaches, visual disturbances, galactorrhea, decreased libido, erectile dysfunction, as well as evaluation for visual field defects. MRI of the pituitary can further characterize the lesion as either a macroadenoma, 1 cm or larger in its greatest dimension, or a microadenoma, less than 1 cm or normal imaging. Approximately one-third of men with hyperprolactinemia are found to have macroadenomas of the pituitary, which are more likely to lead to mass effect symptoms, such as headaches or visual field defects.
Treatment of hyperprolactinemia depends on the size of the lesion. Microadenomas are treated with dopamine agonists, which suppress the production of prolactin. Cabergoline has become the standard of care for such lesions with superior efficacy and a more acceptable side effect profile when compared with bromocriptine. Traditionally, the presence of a macroadenoma was indication for surgical resection. Advances in surgical and endoscopic approaches to resection have led to decreased invasiveness and potential morbidity from this surgery. Despite this, some macroadenomas are initially treated medically with surgical excision reserved for patients who fail treatment. With treatment, patients have been shown to have normalized testosterone levels, improved libido, and better erectile function, as well as improvements in sperm count.
Congenital Adrenal Hyperplasia
Congenital adrenal hyperplasia is caused by a variety of enzyme defects in the steroidogenesis pathway, with 21-hydroxylase deficiency accounting for the majority of cases. The resultant downstream deficiency of cortisol production causes the pituitary to release excessive adrenocorticotropic hormone, which further stimulates the adrenal gland, leading to hyperplasia. As a result of the deficient steroidogenic pathway, an excess of adrenal androgens is produced. These androgenic hormones suppress pituitary release of gonadotropins, which leads to decreased testicular production of testosterone and impaired fertility. The phenotype of men with congenital adrenal hyperplasia is very variable and depends on the degree of androgen excess. Some congenital adrenal hyperplasia patients with a mild phenotype are capable of achieving pregnancy with no treatment, whereas others require treatment to maximize their fertility potential.
Hyperestrogenemia
Estrogen is an important negative feedback signal in the hypothalamic–pituitary–gonadal (HPG) axis. Testosterone, produced by the testis, is converted to estradiol by aromatase in peripheral tissues. Estradiol then acts directly on the hypothalamus and pituitary, via the estrogen receptor, giving a strong inhibitory signal that suppresses the release of GnRH, FSH, and LH. In addition to the HPG axis suppression, estrogen in excess can directly impair spermatogenesis. Men with hyperestrogenemia are found to have low serum FSH, LH, and testosterone in the setting of elevated serum estradiol.
Hyperestrogenemia may be owing to liver failure or tumors that produce estrogen, but it is most commonly caused by obesity. Increased adipose tissue, which contains aromatase, leads to increased conversion of testosterone to estradiol. Aromatase inhibitors can both decrease the hyperestrogenemia and increase testosterone levels. Additionally, weight loss has been shown to improve the testosterone/estrogen balance.
Klinefelter Syndrome
Patients with Klinefelter syndrome have an extra X chromosome (47,XXY) owing to a nondisjunction event that usually happens during meiosis of either the paternal or maternal gamete. These patients often present to the infertility specialist with nonobstructive azoospermia. In fact, up to 10% of patients presenting with nonobstructive azoospermia have Klinefelter syndrome, which makes it the most common genetic cause for nonobstructive azoospermia. The original description by Klinefelter in 1942 included hypergonadotropic hypogonadism, gynecomastia, and infertility. Additionally, the phenotype can include tall stature, decreased virilization, mild cognitive impairment, and small testes. Up to 10% of Klinefelter patients have a mosaic pattern with cells containing either 46,XY or 47,XXY, owing to a mitotic nondisjunction during embryogenesis. For patients with this mosaic pattern, it is possible to have sperm in the ejaculate, albeit typically at severely oligozoospermic levels.
The mechanism by which Klinefelter patients are azoospermic or oligozoospermic is multifactorial, with hypogonadism probably playing a part. Regardless, microsurgical testicular sperm extraction has become the standard of care for Klinefelter patients with sperm retrieval rates ranging as high as 69%. Sperm extraction techniques coupled with in vitro fertilization and intracytoplasmic sperm injection have had high fertilization rates in Klinefelter patients. However, patients must be counseled that the XXY genotype and Klinefelter syndrome may be passed to the offspring. Additionally, preimplantation genetic diagnosis techniques should be considered to limit the risk of Klinefelter syndrome in the offspring.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


