CHAPTER 68 Sclerosing Cholangitis and Recurrent Pyogenic Cholangitis
Dr. Bruce Y. Tung contributed to this chapter in the previous edition of this book.
Sclerosing cholangitis encompasses a spectrum of cholestatic conditions that are characterized by patchy inflammation, fibrosis, and destruction of the intrahepatic and extrahepatic bile ducts. These conditions are typically chronic, progressive disorders in which persistent biliary damage may lead to biliary obstruction, biliary cirrhosis, and hepatic failure, with associated complications. The first description of sclerosing cholangitis is credited to Delbet in 1924.1 Although considered for many years to be an extremely rare disorder, the advent of endoscopic retrograde cholangiopancreatography (ERCP) in the 1970s has allowed an improved understanding of the true prevalence of this disorder and facilitated careful study of its natural history. Nevertheless, many aspects of sclerosing cholangitis remain poorly understood; most notably lacking are a detailed knowledge of its etiology and proven effective medical therapy.
A cholangiographic appearance of diffuse stricturing and segmental dilatation of the biliary system, designated sclerosing cholangitis, may be observed in many distinct conditions. The most frequent is primary sclerosing cholangitis (PSC), an idiopathic disorder that usually occurs in association with inflammatory bowel disease (IBD) but may develop independently. PSC may also be associated with a wide variety of fibrotic, autoimmune, and infiltrative disorders, although whether such associations imply a common pathogenesis or epiphenomena is unclear (Table 68-1). PSC is also associated with various immunodeficiency states; in such cases biliary abnormalities may be caused by infection with an opportunistic pathogen. The term secondary sclerosing cholangitis refers to a clinical and radiologic syndrome that is similar to PSC but develops as a consequence of a known pathogenesis or injury. Obstructive, toxic, ischemic, and neoplastic causes of secondary sclerosing cholangitis have been described (see Table 68-1). This chapter focuses on PSC and recurrent pyogenic cholangitis.
Table 68-1 Classification and Diseases Associated with Sclerosing Cholangitis
Secondary Sclerosing Cholangitis
PRIMARY SCLEROSING CHOLANGITIS
DIAGNOSIS
No standardized criteria for the diagnosis of PSC have been universally adopted. Early diagnostic criteria included diffuse intra- and extrahepatic bile duct strictures occurring in the absence of prior biliary surgery or cholelithiasis and after exclusion of cholangiocarcinoma.2 These criteria were later modified because of the recognition that the clinical spectrum of PSC is broader than initially appreciated, and strict adherence to the original criteria underestimates the prevalence of the disease. It is now apparent that a form of PSC, termed small-duct PSC, involves only the intrahepatic biliary tree, without obvious extrahepatic duct abnormalities.3 In addition, both cholelithiasis and choledocholithiasis may develop as a consequence of PSC, and their presence does not exclude a diagnosis of underlying PSC.4,5 Furthermore, cholangiocarcinoma is a relatively common complication of PSC, and both conditions frequently coexist.6
The diagnosis of PSC is based on typical cholangiographic findings in the setting of consistent clinical, biochemical, serologic, and histologic findings as well as exclusion of secondary causes of sclerosing cholangitis. The characteristic cholangiographic findings are multifocal stricturing and ectasia of the biliary tree. Areas of narrowing are interspersed with areas of normal or near-normal caliber and of post-stenotic dilatation. Although the majority of patients with PSC have coexisting abnormalities of the intra- and extrahepatic bile ducts, a small percentage have an isolated lesion. Patients with small-duct PSC may have a normal cholangiogram. Gallbladder abnormalities, including tumors, may exist in up to 41% of patients with PSC.7
ERCP is considered the standard for establishing a diagnosis of PSC but carries a risk for complications of up to 10% in patients with PSC.8,9 Magnetic resonance cholangiopancreatography (MRCP) has largely replaced ERCP for diagnostic cholangiography as a result of improvements in image quality and the noninvasive nature of MRCP (Fig. 68-1). In the few studies that have compared MRCP and ERCP in patients with PSC, MRCP has demonstrated comparable sensitivity for the detection of biliary structuring,10–13 although performance and interpretation of magnetic resonance cholangiograms vary with the technique and institution. ERCP has the advantage of combining high-resolution cholangiography with the potential for advanced diagnostic and therapeutic interventions, including brush cytology or intraductal biopsy for the diagnosis of cholangiocarcinoma, balloon or catheter dilation of strictures, biliary stent placement, sphincterotomy, and stone removal. Percutaneous transhepatic cholangiography (THC) may also yield diagnostic images and allow therapeutic intervention but requires percutaneous puncture and may be technically difficult if the intrahepatic bile ducts are not sufficiently dilated (see Chapter 70).
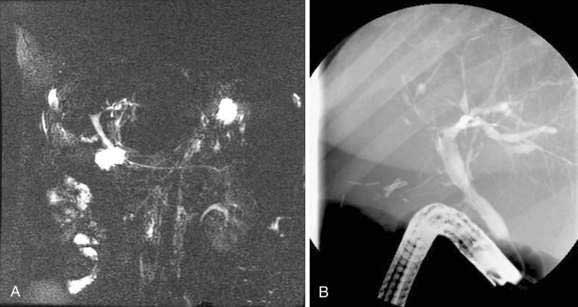
Patients with IBD and a cholestatic pattern of liver biochemical test elevations should undergo imaging of the hepatobiliary system because of the relatively high pretest probability of PSC. Ultrasonography or computed tomography (CT) may be useful for planning further diagnostic and therapeutic strategies in selected patients, but they are usually insufficient for a diagnosis of PSC because normal findings do not exclude the diagnosis. The decision as to which method of cholangiography to perform must be individualized. In most cases, ERCP is the initial test of choice for patients in whom a therapeutic intervention or the need for brush cytology is anticipated. In an asymptomatic patient with mild liver biochemical abnormalities who is unlikely to require therapeutic intervention, MRCP is the preferred initial test if the images are reliable.13 When MRCP is nondiagnostic and clinical suspicion for PSC remains, diagnostic ERCP is indicated.
Differential Diagnosis
In a patient with a cholangiographic appearance characteristic of sclerosing cholangitis, secondary causes of sclerosing cholangitis must be excluded (see Table 68-1). Patients with the acquired immunodeficiency syndrome (AIDS) and a CD4+ T-lymphocyte count below 100/mm3 can exhibit a cholangiographic appearance identical to that of PSC; this entity is termed AIDS cholangiopathy. Cryptosporidium, Microsporidium, cytomegalovirus, and other organisms have been isolated from the bile of affected patients.14,15 Exposure of the bile ducts to toxins such as intra-arterial floxuridine (FUDR)16 and formaldehyde administered to treat a hydatid cyst, when the cyst communicates with the biliary tract,17 can produce a similar cholangiographic appearance.
Primary biliary cirrhosis (PBC) is another chronic cholestatic condition that shares some clinical features with PSC (see Chapter 89); however, PBC predominantly affects middle-aged women, has no association with IBD, and is associated strongly with high titers of antimitochondrial antibodies. Whereas liver histologic findings in the two disorders overlap substantially,18 the distinction between the two is readily apparent on cholangiography. Patients with advanced PBC may demonstrate smooth tapering and narrowing of the intrahepatic bile ducts, but ductal irregularity or strictures are not seen and extrahepatic lesions do not occur. Antimitochondrial antibody-negative PBC (autoimmune cholangitis) may be difficult to distinguish from small-duct PSC because serologic profiles and cholangiographic findings may overlap, but the demographic and histologic features of the two disorders are distinct (see Chapter 89).
Autoimmune hepatitis may also be difficult to distinguish from PSC (see Chapter 88). In the pediatric population, PSC typically manifests with features of autoimmune hepatitis, and cholangiography is necessary to distinguish the two disorders (see Chapter 62).19 With use of a standardized scoring system for the diagnosis of autoimmune hepatitis, 7.5% of patients with PSC are characterized as “definite” or “probable” for the diagnosis of autoimmune hepatitis, thereby underscoring the need for cholangiography when PSC is suspected.20 Features suggestive of autoimmune hepatitis include female predominance, a hepatocellular rather than cholestatic pattern of liver biochemical test abnormalities, hypergammaglobulinemia, high titers of antinuclear and anti-smooth muscle antibodies, histologic evidence of periportal necroinflammation, and clinical response to glucocorticoid therapy. An overlap syndrome between PSC and autoimmune hepatitis has been described; it consists of a mixed cholestatic and hepatocellular pattern of liver biochemical test abnormalities, the presence in serum of autoantibodies including antineutrophil cytoplasmic antibodies (ANCA), cholangiography consistent with PSC, and histologic evidence of periductular fibrosis as well as periportal necroinflammation.21,22
A disorder of the pancreaticobiliary tree termed autoimmune pancreatitis, sclerosing pancreatocholangitis, or immunoglobulin (Ig) G4–associated cholangitis has been described.23 This disorder shares cholangiographic and clinical features with PSC but differs in its responsiveness to glucocorticoid therapy. Serum levels of IgG4 are often elevated in this disorder, and high numbers of IgG4 positive lymphocytes (>20 per high-powered field) are identified in pinch biopsies obtained from the major papilla or bile duct and may be diagnostic. Although specific diagnostic criteria for this disorder are still emerging, persons without IBD who present with symptoms and cholangiographic findings consistent with PSC should undergo measurement of serum IgG4 levels as well as endoscopy and biopsy of the major papilla to exclude IgG4-associated cholangitis (see Chapter 59).23–25
EPIDEMIOLOGY
Determination of the true incidence and prevalence of PSC is complicated by the variable presentation of the disease, inconsistent diagnostic criteria, and referral bias inherent in many published studies. Two population-based studies have provided the most accurate epidemiologic estimates of PSC in Western populations. On the basis of these studies performed in the United States and Norway, the incidence of PSC is estimated to be 0.9 to 1.3 per 100,000, and the point prevalence is estimated to be 8.5 to 13.6 per 100,000.26,27
Although PSC has been diagnosed in neonates and as late as the eighth decade of life, most patients present between the ages of 25 and 45 years, with a mean age of approximately 39 years.19,28–33 Approximately 70% of patients with PSC are men,27–31 but in the subset of patients without IBD, the male-to-female ratio is lower (0.72:1).34 Women with PSC are generally older at diagnosis.27,35 PSC is also associated with nonsmoking, an effect that cannot be explained entirely by the association between ulcerative colitis (UC) and nonsmoking.36,37
PRIMARY SCLEROSING CHOLANGITIS AND INFLAMMATORY BOWEL DISEASE
The relationship between PSC and IBD is striking and incompletely understood. Approximately 80% of all patients with PSC have concomitant IBD.27–29,31,38,39 Conversely, PSC is present in 2.4% to 4.0% of all patients with chronic UC and 1.4% to 3.4% of patients with Crohn’s disease.35,38,40,41 Of patients with both PSC and IBD, approximately 85% to 90% have UC and the remainder have Crohn’s colitis or ileocolitis. The association with IBD is stronger with more extensive colonic involvement; the prevalence of PSC is approximately 5.5% in those with pancolitis, in contrast to 0.5% in those with only distal colitis.35 PSC is not thought to occur in association with Crohn’s disease isolated to the small intestine. Racial differences in the association between PSC and IBD may exist; concomitant IBD is seen in only 21% of Japanese patients with PSC.42
Despite the strong association between PSC and UC, the two diseases often progress independently of each other.43 Although IBD is typically diagnosed before PSC, UC may be newly diagnosed years after liver transplantation for end-stage liver disease caused by PSC. Conversely, PSC may be diagnosed years after total proctocolectomy for UC.44,45
Whether PSC differs clinically in patients with and without concomitant IBD is unclear. Older reports demonstrated no histologic46 or cholangiographic47 differences between patients with or without IBD. One study,34 however, suggested that patients without IBD are more likely to be female, have disease isolated to the extrahepatic ducts, and be symptomatic at the time of diagnosis. Of the multiple multivariate analyses performed to identify risk factors for progression of PSC (see later), only one found that the presence of IBD has a significant independent effect on progression of PSC.29 Some patients without overt IBD may have subclinical histologic changes detected in the colon or may develop overt colitis at a later date.43 Therefore, a high index of suspicion for the emergence of IBD is warranted, and colonoscopy with random biopsies of the colonic mucosa is recommended for all patients with a new diagnosis of PSC.
ETIOLOGY AND PATHOGENESIS
Genetic Factors
The importance of genetic factors in the pathogenesis of PSC is demonstrated by familial occurrence of the disease and its associations with specific human leukocyte antigen (HLA) haplotypes. Although uncommon, familial clustering of cases of PSC have been reported.48,49 Furthermore, PSC is strongly associated with specific HLA haplotypes. Early studies described an overrepresentation of HLA B8 and DR3 in patients with PSC; these haplotypes are also associated with other autoimmune disorders such as myasthenia gravis and autoimmune hepatitis.50,51 These findings are not explained simply by the association between PSC and IBD because HLA B8 and DR3 are not overrepresented in patients with IBD but without PSC. The subsequent development of molecular genotyping demonstrated that the most common allele in patients with PSC is DRB3*0101, which encodes the DRw52a antigen. One study found this allele in 100% of 29 patients with PSC who underwent liver transplantation,52 but subsequent studies have demonstrated this allele in only 50% to 55% of patients with PSC.53–55 Currently, the extended HLA haplotypes that are most strongly associated with PSC are as follows:53,55,56
The strongest association maps to the HLA class I/III boundary on chromosome 6p21. Strong disease associations have been identified with the MICA*008 allele57 and the tumor necrosis factor α-2 allele.58,59 Despite the multiple HLA associations described, however, a single HLA-encoded gene that determines susceptibility to PSC appears unlikely. More likely are multiple HLA susceptibility loci, which may explain in part why PSC is a relatively rare disease even though the HLA haplotypes associated with PSC are relatively common in populations of Northern European descent.
Also controversial is whether specific haplotypes are associated with disease outcomes. One study suggested a poor prognosis in patients with PSC and HLA DR4,60 but this finding was not confirmed.55 A more recent multicenter study involving 256 patients with PSC showed that the heterozygous haplotype DR3,DQ2 was associated with a greater risk of liver transplantation or death and the DQ6 haplotype was associated with a decreased risk of disease progression.61
The relationship between several non-major histocompatibility complex (MHC) genes and susceptibility to PSC has also been investigated. An initial study reported an association with polymorphisms in the gene encoding matrix metalloproteinase 3 (MMP-3) and postulated a role for MMP-3 in progression of PSC because of its ability to regulate fibrosis and immune activation.62 A subsequent report, however, did not confirm an association between either MMP-1 or MMP-3 polymorphisms and PSC.63 Similarly, no associations between PSC and polymorphisms in the interleukin (IL)-1 or IL-10 genes have been noted.64
Immunologic Factors
Evidence suggests that the immune system plays a key role in the etiology and pathogenesis of PSC, including the multiple associations between PSC and other autoimmune disorders. The most frequently associated autoimmune disorders include type I diabetes mellitus and Graves’ disease, which are more common in patients with PSC and IBD than in patients with IBD alone.65 In addition, as described earlier, an overlap syndrome that includes features of both PSC and autoimmune hepatitis has been described.21,22,66 In rare cases, well-characterized autoimmune hepatitis may evolve into sclerosing cholangitis, suggesting that both diseases may be part of the same clinical spectrum.67 Unlike most other autoimmune disorders, however, PSC has an approximately 2:1 male predominance, is not associated with disease-specific autoantibodies, and does not exhibit a consistent clinical response to immunosuppressive therapy.
A wide range of serum autoantibodies are found in patients with PSC, although none is specific for the disease. Whether any of these associated antibodies plays a key role in the pathogenesis of the disease process or whether they represent simple epiphenomena is unclear. Antinuclear antibodies may be present in 24% to 53%, anti-smooth muscle antibodies in 13% to 20%, and an atypical perinuclear ANCA (pANCA) in 65% to 88% of patients with PSC.68–75 Antibodies directed against cardiolipin, bactericidal/permeability-increasing protein, cathepsin G, and lactoferrin have also been detected.74–75 Antibodies directed against an epitope shared by colonic and biliary epithelial cells have been demonstrated and may suggest a mechanism for the association between IBD and PSC.76 Autoantibodies that bind to human biliary epithelial cells (anti-BEC) have been shown to induce expression of IL-6 and the cell adhesion molecule CD44; this finding could represent a potential mechanism for the inflammatory bile duct destruction seen in patients with PSC.77
Abnormalities of both humoral and cellular immunity have been described in patients with PSC. They include an increase in circulating immune complexes, deficient clearance of immune complexes, and activation of the classical pathway of the complement system.78–80 Serum elevations of IL-8 and IL-10 also suggest exaggerated humoral immunity.81 Some of the abnormalities in cellular-mediated immunity that have been described include a decrease in circulating CD8+ cytotoxic T cells,82 increased numbers of γδ T cells in peripheral blood as well as portal areas of the liver,83 and overrepresentation of Vβ3 T-cell receptor gene segments in hepatic (but not peripheral) T-cell populations.84
Biliary Epithelial Cells
The role of biliary epithelial cells in the pathogenesis of PSC remains unclear. Biliary epithelial cells could serve as a trigger and a target for immune-mediated injury. Biliary epithelial cells have been shown to express MHC class II antigens85 and adhesion molecules such as intracellular adhesion molecule-1 (ICAM-1)86 and could play a role as antigen-presenting cells to T lymphocytes. The expression of these molecules can be regulated on biliary epithelial cells by various cytokines, including IL-2 and interferon-γ.87 Biliary epithelial cells, however, may not express the co-stimulatory ligands necessary for activation of T lymphocytes.88 In addition, many of the same findings are seen in patients with PBC and extrahepatic bile duct obstruction as well, making it less likely that they play a primary pathogenic role in PSC.85
Infectious and Toxic Factors
The strong association between PSC and colitis has provoked the theory that penetration of infectious or toxic agents through an inflamed colon into the portal system may play an important role in the pathogenesis of PSC. Bile culture results have been positive in explanted livers in a majority of patients with PSC, although the number of bacterial strains has correlated inversely with the time since the last endoscopic intervention.89 In addition, bacterial endotoxin has been shown to accumulate in biliary epithelial cells in patients with PSC and PBC.90 In patients with AIDS cholangiopathy, a variety of organisms, including Cryptosporidium, Microsporidium, and cytomegalovirus, have been isolated from the bile.14,15 A study that evaluated serologic profiles in 41 patients with PSC found a higher percentage with Chlamydia lipopolysaccharide antibodies than in a large control population. No association was seen with any other microorganisms, including Mycoplasma and 22 viruses tested.91 Further study is necessary before a direct link between PSC and Chlamydia, or any other infectious agent, can be established.
A loss of normal colonic mucosal barrier because of inflammation could allow portal inflow of noninfectious toxins. Toxic damage leading to sclerosing cholangitis has been demonstrated in humans as well as animal models. Biliary exposure to caustic agents17 or hepatic artery infusion of chemotherapeutic agents such as FUDR16 can produce a cholangiographic appearance identical to that of PSC. In a rat model, administration of the biliary toxin α-naphthylisothiocyanate led to the development of a chronic cholangitis similar to sclerosing cholangitis in humans.92 The toxic injury hypothesis, however, does not explain why PSC is not associated with the severity of colonic inflammation in patients with IBD and why PSC may develop years after a patient has undergone total proctocolectomy.
Vascular Factors
Ischemia has been postulated to play a role in the pathogenesis of PSC because a similar cholangiographic appearance may be found after surgical trauma to the biliary vascular supply93 and after hepatic artery thrombosis or arterial fibrointimal hyperplasia after liver transplantation.94,95 In addition, PSC is associated with the presence of autoantibodies such as pANCA and anti-cardiolipin antibodies. These autoantibodies, in turn, are strongly associated with vasculitides such as Wegener’s granulomatosis, polyarteritis nodosa, and thrombotic syndromes. These associations suggest that immune-mediated vascular injury plays a role in the pathogenesis of PSC.
NATURAL HISTORY AND PROGNOSTIC MODELS
PSC is typically a progressive disease, although the natural history is incompletely understood.29–31,96–98 The disease may be considered to progress through the following four clinical phases, although some phases may not develop or be apparent in an individual patient:
Asymptomatic Primary Sclerosing Cholangitis
Asymptomatic patients with PSC make up 15% to 44% of cohorts examined in published studies29,31,32,98,99 Some reports have suggested that asymptomatic patients typically have a benign course of disease. Helzberg and colleagues98 reported on 11 asymptomatic patients with PSC who were followed for a mean of 37 months, and all 11 remained asymptomatic without evidence of progressive disease. By contrast, Porayko and colleagues99 followed 45 asymptomatic patients with PSC for a median of 6.25 years, and during the surveillance period, liver failure, resulting in liver transplantation or death, developed in 13 (31%). Overall, symptoms developed in 24 (53%), and progressive liver disease, demonstrated by new symptoms or signs, worsening cholangiographic findings, or progressive liver histologic abnormalities, developed in 34 (76%) patients. The Kaplan-Meier estimate of median survival free of liver failure in this study was 71% at seven years for the asymptomatic patients, significantly lower than the 96% expected on the basis of an age-, sex-, and race-matched U.S. control population. Differences in the rates of progression between these studies98,99 may be the result of differences in patient populations, the definition of “asymptomatic,” and the duration of clinical follow-up.
Symptomatic Primary Sclerosing Cholangitis
Patients with symptoms at the time of diagnosis generally have a worse prognosis than asymptomatic patients.29,30 The clinical stage is likely more advanced at the time of diagnosis in symptomatic patients, who have more severe biochemical derangements, more abnormalities on cholangiography, and a higher histologic stage on liver biopsy specimens than asymptomatic patients. Wiesner and colleagues29 compared the natural history of PSC in 37 asymptomatic patients with that in 137 patients who were symptomatic at the time of diagnosis. After a mean follow-up of six years, 55 (40%) of the symptomatic patients had died, compared with 4 (11%) in the asymptomatic group. The Kaplan-Meier estimate of median survival for the entire cohort was 11.9 years; for the symptomatic cohort, the estimated median survival was between 8 and 9 years. Farrant and colleagues30 described the natural history of PSC in 126 patients, of whom 84% were symptomatic. After a median follow-up of 5.8 years, the estimated median survival was 12 years. Similar findings were reported in a large study by Broome and colleagues.31 In 305 patients with PSC followed for a median of 5.25 years, of whom 44% were asymptomatic, the estimated median survival was 12 years. Patients who were symptomatic at the time of entry into the study had a significantly worse expected survival (9.3 years) than asymptomatic patients. A study of 174 patients with PSC by Ponsioen and colleagues97 suggested a better overall prognosis, with a median expected survival of 18 years. The reason for improved survival in this most recent study is not known, but patient data were predominantly from the 1990s, compared with data from the 1970s and 1980s in the other studies described. Although therapeutic advances were not dramatic in the interim, earlier diagnosis in the 1990s may have led to differences in patient selection that appeared to affect outcomes.
Small-Duct Primary Sclerosing Cholangitis
Patients who have histologic, biochemical, and clinical features of PSC but a normal cholangiogram are considered to have small-duct PSC, which accounts for 5% to 20% of all patients with PSC.3,100 Three studies have performed extended clinical follow-up in patients with small-duct PSC.100–102 In these studies, 12% to 17% of patients progressed to classic large-duct PSC over long-term follow-up, although the true rate may be higher because cholangiograms were not obtained routinely in all patients. Cholangiocarcinoma did not develop in any patient over a median follow-up of 63 to 126 months, and survival in the small-duct PSC group was better than that of matched control groups with classic PSC.100–102 Therefore, small-duct appears to represent an early stage of PSC, may progress to large-duct PSC in a small percentage of patients, and is associated with a better prognosis than classic PSC.
Prognostic Models
Multivariable prognostic models that have been developed to predict survival in patients with PSC are shown in Table 68-2. In an early multivariable analysis, hepatomegaly and a serum bilirubin level > 1.5 mg/dL were found to be independently associated with a poor prognosis in PSC. The patient’s age, histologic findings, presence of concomitant IBD, and pattern of cholangiographic involvement did not correlate independently with survival in this study.98 Wiesner and colleagues29 developed a prognostic model based on age, serum bilirubin level, hemoglobin value, presence or absence of IBD, and histologic stage. With this model, three risk groups (low, intermediate, high) were formed, and predicted survival curves were shown to be similar to observed survival curves. Farrant and colleagues30 developed a multivariable prognostic model in which hepatomegaly, splenomegaly, serum alkaline phosphatase level, histologic stage, and age at presentation were found to be important independent factors. Dickson and colleagues103 then presented a model developed from a multicenter collaboration in which data from 426 patients with PSC were pooled. In this analysis, the patient’s age, serum bilirubin level, histologic stage, and presence of splenomegaly were found to correlate independently with survival, and the model was validated against the observed survival data in a subgroup of the entire cohort. Broome and colleagues31 found the patient’s age, histologic stage, and serum bilirubin level to be independent predictors of survival in 305 patients with PSC, but this prognostic model was not validated independently. Most recently, Kim and colleagues,104 using easily obtainable clinical and biochemical factors, revised an earlier predictive model that did not require liver biopsy and did not rely on subjective physical findings such as splenomegaly or hepatomegaly. This revised natural history model (revised Mayo risk model) found the patient’s age, serum bilirubin level, serum aspartate aminotransferase (AST) level, and serum albumin level and a history of variceal bleeding to be independent predictors of survival. The model was generated from data on 529 patients from 5 centers and was validated using data from another center that had not been used in the development of the model.
Table 68-2 Independent Predictors of Survival and Prognostic Index Formulas Used in Natural History Models of Primary Sclerosing Cholangitis*

The Child (Child-Pugh) classification may also be used to predict survival in patients with PSC (see Chapter 90). Shetty and colleagues105 found that Kaplan-Meier seven-year survival rates for patients with Child class A, B, and C cirrhosis caused by PSC were 89.8%, 68%, and 24.9%, respectively. Subsequent evaluation, however, suggested that the Child classification is less accurate than the revised Mayo risk model, especially for patients with early-stage PSC.106
CLINICAL FEATURES
Symptoms
The initial clinical presentation of PSC can be quite varied and may run the gamut from asymptomatic elevations of serum alkaline phosphatase levels to decompensated cirrhosis with jaundice, ascites, hepatic encephalopathy, or variceal bleeding. The most common symptoms at the time of presentation include jaundice, fatigue, pruritus, and abdominal pain.19,28–33,107,108 Other associated symptoms may include fever, chills, night sweats, and weight loss (Table 68-3). The onset of these symptoms is typically insidious, although an acute hepatitis-like presentation has been described.40 Increasingly, PSC is diagnosed in an asymptomatic or minimally symptomatic stage. Large series have shown that 15% to 44% of patients with PSC are asymptomatic at the time of diagnosis,29,31,32,98,99 probably because of the routine liver biochemical screening in patients with IBD, as well as the widespread availability of MRCP and ERCP for evaluating elevated serum alkaline phosphatase levels.
Table 68-3 Most Common Symptoms and Signs at the Time of Diagnosis of Primary Sclerosing Cholangitis
| Symptoms | Rate (%) |
| Fatigue | 65-75 |
| Abdominal pain | 24-72 |
| Pruritus | 15-69 |
| Fever/night sweats | 13-45 |
| Asymptomatic | 15-44 |
| Weight loss | 10-34 |
| Signs | |
| Jaundice | 30-73 |
| Hepatomegaly | 34-62 |
| Splenomegaly | 32-34 |
| Hyperpigmentation | 14-25 |
| Ascites | 4-7 |
Data from references 19, 29–33, 98, 99, 107, 108.
Symptoms of PSC are often intermittent. Episodes of pruritus, jaundice, abdominal pain, and fever are typically interspersed with asymptomatic periods of varying duration.39,107,108 The intermittency of the symptoms is thought to reflect intermittent biliary obstruction caused by microlithiasis and sludge.5,109 This obstruction may predispose to cholestasis and induce an acute inflammatory reaction. Secondary bacterial infection may result in low-grade cholangitis and predispose to pigment stone formation.5
Physical Examination
Physical findings may be normal in patients with PSC, particularly those who are asymptomatic. When physical abnormalities are present, the most common include hepatomegaly, jaundice, and splenomegaly (see Table 68-3). Skin findings are common and include cutaneous hyperpigmentation, excoriations resulting from pruritus, and xanthomata. As liver disease progresses, spider angiomas, muscular atrophy, peripheral edema, ascites, and other signs of advanced liver disease may appear.28–30
Laboratory Findings
Chronic elevation of serum alkaline phosphatase levels, typically three to five times normal, is the biochemical hallmark of PSC. A normal alkaline phosphatase level, however, may be found in up to 6% of patients with cholangiographically proved PSC.110,111 In some cases, an advanced histologic stage has been demonstrated on a liver biopsy specimen despite normal serum alkaline phosphatase levels.110 Serum aminotransferase levels are typically elevated, although rarely above four to five times normal except in the pediatric population.112 The serum bilirubin level may be normal or elevated and often fluctuates. When the serum bilirubin level is elevated, the bilirubin is predominantly conjugated. Reductions in the serum albumin level and prolongation of the prothrombin time may reflect hepatic synthetic dysfunction with advanced liver disease. In addition, malnutrition and underlying IBD may lower serum albumin levels. Vitamin K malabsorption related to cholestasis may play a role in prolonging the prothrombin time. Other nonspecific consequences of cholestasis are elevations in serum copper, serum ceruloplasmin, and hepatic copper levels, increased urinary copper excretion, and elevated serum cholesterol levels.
Several immunologic markers and serum autoantibodies are found in the majority of patients with PSC, although none is specific for the disease. Hyperglobulinemia is frequent; serum IgM levels are elevated in up to 50% of patients, and IgG and IgA levels also may be elevated.28,29,28,107 Antinuclear antibodies, often in low titer, may be detected in 24% to 53% of patients. Anti-smooth muscle antibodies are found in 13% to 20% of patients, but antimitochondrial antibodies are found in less than 10%.26,28,70,73,75 Most commonly found in patients with PSC are pANCA,72 which are detected in 65% to 88% of patients and appear to react to a heterogeneous group of antigens.70,71,74,75 These antigens have been found to represent neutrophil nuclear envelope proteins predominantly, and the corresponding antibodies have been referred to as “antineutrophil nuclear antibodies” (ANNA).113 In contrast to Wegener’s granulomatosis, titers of pANCA do not appear to correlate with disease activity, severity, or response to medical therapy in patients with PSC.72 Furthermore, the presence of autoantibodies does not appear to differ in patients with and without IBD. Anti-cardiolipin antibodies are detected in 66% of patients with PSC, and the titer has been reported to correlate with disease severity.75 In general, despite the high frequency of autoantibodies in patients with PSC, a clear association between the presence of these antibodies, pathogenesis of the disease, and prognosis or response to treatment remains unproved. Measurement of autoantibodies is therefore of limited clinical value in patients with PSC.
Imaging Findings
Cholangiography by ERCP, MRCP, or percutaneous THC establishes a diagnosis of PSC and provides information regarding the distribution and extent of disease. The characteristic cholangiographic findings include multifocal stricturing and ectasia of the biliary tree. Areas of narrowing are interspersed with areas of normal or near-normal caliber and areas of post-stenotic dilatation. The result is a classic “beaded” appearance to the biliary tree. The strictured segments are usually short, annular, or band-like in appearance (Fig. 68-2), although longer confluent strictures may be seen in more advanced disease. Localized segments of dilated ducts may have a saccular or diverticular appearance. Major areas of focal, tight narrowing known as dominant strictures may be seen and often involve the bifurcation of the hepatic duct.114 At times, diffuse involvement of the intrahepatic biliary tree may give a pruned appearance that is difficult to distinguish from the diffuse intrahepatic duct attenuation seen in patients with cirrhosis of any cause; irregularity of the duct wall or concomitant involvement of the extrahepatic bile duct supports a diagnosis of PSC.
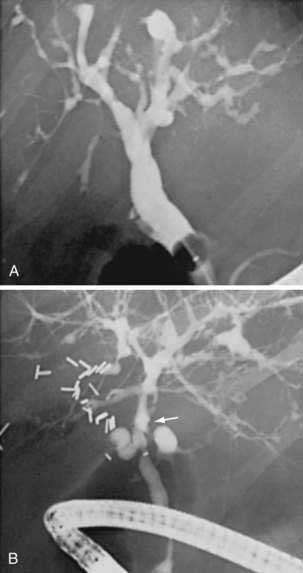
Both the extrahepatic and intrahepatic bile ducts are abnormal in approximately 75% of cases. The intrahepatic ducts alone may be involved in 15% to 20% of cases.28,31,35,107 Abnormalities of the extrahepatic biliary tree in the absence of intrahepatic involvement are less common.98,99 The cystic duct and gallbladder may be involved in up to 15% of patients but may not be well visualized on routine cholangiography.115 Pancreatic duct irregularities similar to those seen in chronic pancreatitis may rarely be noted.116
PATHOLOGY
Gross and histologic specimens from the extrahepatic bile ducts demonstrate a diffusely thickened, fibrotic duct wall. The fibrosis is accompanied by a mixed inflammatory infiltrate that may involve the epithelium and biliary glands.117,118 Florid hyperplasia of the biliary glands with accompanying neural proliferation has been described.119 Examination of PSC explants removed at the time of liver transplantation has demonstrated areas of thin-walled saccular dilatation, termed “cholangiectasias,” that correspond to the beaded appearance on cholangiography.120
A wide range of liver biopsy findings may be seen in patients with PSC. For this reason, histologic findings are not typically diagnostic for PSC. The characteristic bile duct lesion is a fibro-obliterative process that may lead to an “onionskin” appearance of concentric fibrosis surrounding medium-sized bile ducts (Fig. 68-3); however, this finding is seen in less than one half of biopsy specimens.117,121,122 The smaller interlobular and septal bile duct branches may be entirely obliterated by this process, resulting in fibro-obliterative cholangitis. This finding is present in only 5% to 10% of biopsy specimens but is thought to be virtually pathognomonic of PSC.121 In this process, the biliary epithelium may degenerate and atrophy and be replaced entirely by fibrous cords. Other characteristic histopathologic findings may include bile duct proliferation, periductal inflammation, and ductopenia. The degree of inflammation can be quite variable but is typically a portal-based mixture of lymphocytes, plasma cells, and neutrophils with a periductal focus. Lymphoid follicles or aggregates may also be seen.121,122
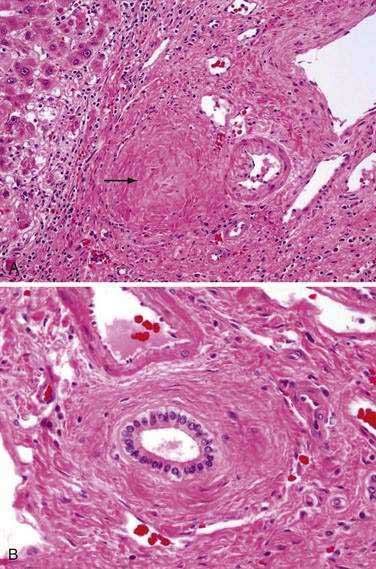
The histologic progression of PSC can be divided into four stages, analogous to a similar staging system in PBC46 (See Chapter 89). In stage I (portal stage) changes are confined to the portal tracts and consist of portal inflammation, connective tissue expansion, and cholangitis. Stage II (periportal stage) is characterized by expansion of inflammatory and fibrotic processes beyond the confines of the limiting plate, resulting in “piecemeal necrosis” (interface hepatitis) and periportal fibrosis. Depending on the degree of biliary obstruction, ductular proliferation and cholangitis may be of varying severity. Stage III (septal stage) is characterized by fibrous septa that bridge from one portal tract to the next. Bridging necrosis may occasionally be seen but is uncommon. Stage IV (cirrhotic stage) implies progression to biliary cirrhosis. The degree of inflammatory activity may subside as the stage of disease progresses, and focal bile ductular proliferation may be striking. A study that examined the time course of progression through the histologic stages revealed that for patients with stage II disease, 42%, 66%, and 93% progressed to a higher histologic stage at one, two, and five years, respectively.123 For patients initially with stage II disease, progression to biliary cirrhosis (stage IV) occurred in 14%, 25%, and 52%, respectively. Regression of stage was observed in 15% of patients and probably reflected sampling variability in the histologic assessment.
Many of the histologic findings of PSC are nonspecific and may be seen in other disorders. In particular, the histologic distinction between PSC and PBC may be difficult to discern. In one study, histologic examination could classify only 28% of patients who had one of the two diseases.18 When lymphocytic interface hepatitis is prominent, the distinction from autoimmune hepatitis may be challenging, especially because hypergammaglobulinemia and autoantibodies may be present in both conditions. In addition, an overlap syndrome with features of both PSC and autoimmune hepatitis has been described.21,22,66 When severe cholestasis develops, hepatic copper accumulation can be dramatic and may mimic that seen in Wilson disease.124









